Sequana documentation
Current version: 0.16.11, Mar 18, 2024
SEQUANA


- How to cite:
Citations are important for us to carry on developments. For Sequana library (including the pipelines), please use
Cokelaer et al, (2017), 'Sequana': a Set of Snakemake NGS pipelines, Journal of Open Source Software, 2(16), 352, JOSS DOI doi:10.21105/joss.00352
For the genome coverage tool (sequana_coverage): Desvillechabrol et al, 2018: detection and characterization of genomic variations using running median and mixture models. GigaScience, 7(12), 2018. https://doi.org/10.1093/gigascience/giy110
For Sequanix: Desvillechabrol et al. Sequanix: A Dynamic Graphical Interface for Snakemake Workflows Bioinformatics, bty034, https://doi.org/10.1093/bioinformatics/bty034 Also available on bioRxiv (DOI: https://doi.org/10.1101/162701)
Sequana includes a set of pipelines related to NGS (new generation sequencing) including quality control, variant calling, coverage, taxonomy, transcriptomics. We also ship Sequanix, a graphical user interface for Snakemake pipelines.
name/github |
description |
Latest Pypi version |
Test passing |
apptainers |
|---|---|---|---|---|
Create and Manage Sequana pipeline |
Not required |
|||
Set of wrappers to build pipelines |
Not on pypi |
Not required |
||
Demultiplex your raw data |
License restriction |
|||
denovo sequencing data |
||||
Get Sequencing Quality control |
||||
Map sequences on target genome |
||||
Map sequences on target genome |
||||
Merge barcoded (or unbarcoded) nanopore fastq and reporting |
||||
Pacbio quality control |
||||
Find ribosomal content |
||||
RNA-seq analysis |
||||
Variant Calling |
||||
Coverage (mapping) |
||||
Long read Amplicon Analysis |
||||
reverse complement of sequence data |
||||
downsample sequencing data |
Not required |
|||
remove/select reads mapping a reference |
name/github |
description |
Latest Pypi version |
Test passing |
|---|---|---|---|
Find repeats |
|||
Taxonomy analysis |
Please see the documentation for an up-to-date status and documentation.
Contributors
Maintaining Sequana would not have been possible without users and contributors. Each contribution has been an encouragement to pursue this project. Thanks to all:
Changelog
Version |
Description |
|---|---|
0.17.0 |
|
0.16.9 |
|
0.16.8 |
|
0.16.7 |
|
0.16.6 |
|
0.16.5 |
|
0.16.4 |
|
0.16.3 |
|
0.16.2 |
|
0.16.1 |
|
0.16.0 |
|
0.15.4 |
|
0.15.3 |
|
0.15.2 |
|
0.15.1 |
|
0.15.0 |
|
0.14.6 |
|
0.14.5 |
|
0.14.4 |
|
0.14.3 |
|
0.14.2 |
|
0.14.1 |
|
0.14.0 |
|
0.13.X |
|
0.12.X |
|
What is Sequana ?
Sequana is a versatile tool that provides
A Python library dedicated to NGS analysis (e.g., tools to visualise standard NGS formats).
A set of pipelines dedicated to NGS in the form of Snakefiles (Makefile-like with Python syntax based on snakemake framework).
Original tools to help in the creation of such pipelines including HTML reports.
- Standalone applications:
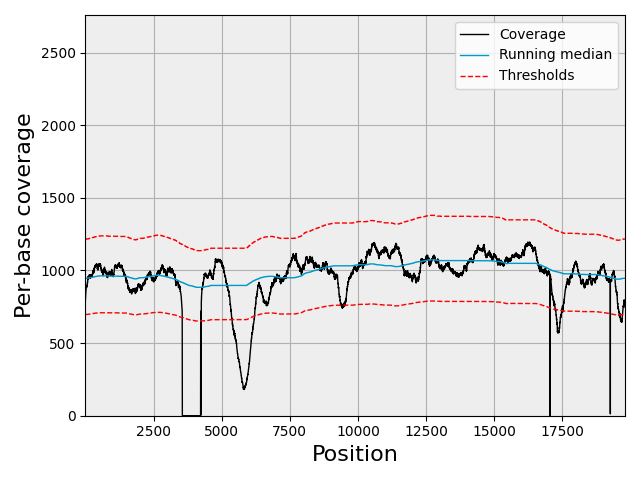
sequana_coverage ease the extraction of genomic regions of interest and genome coverage information
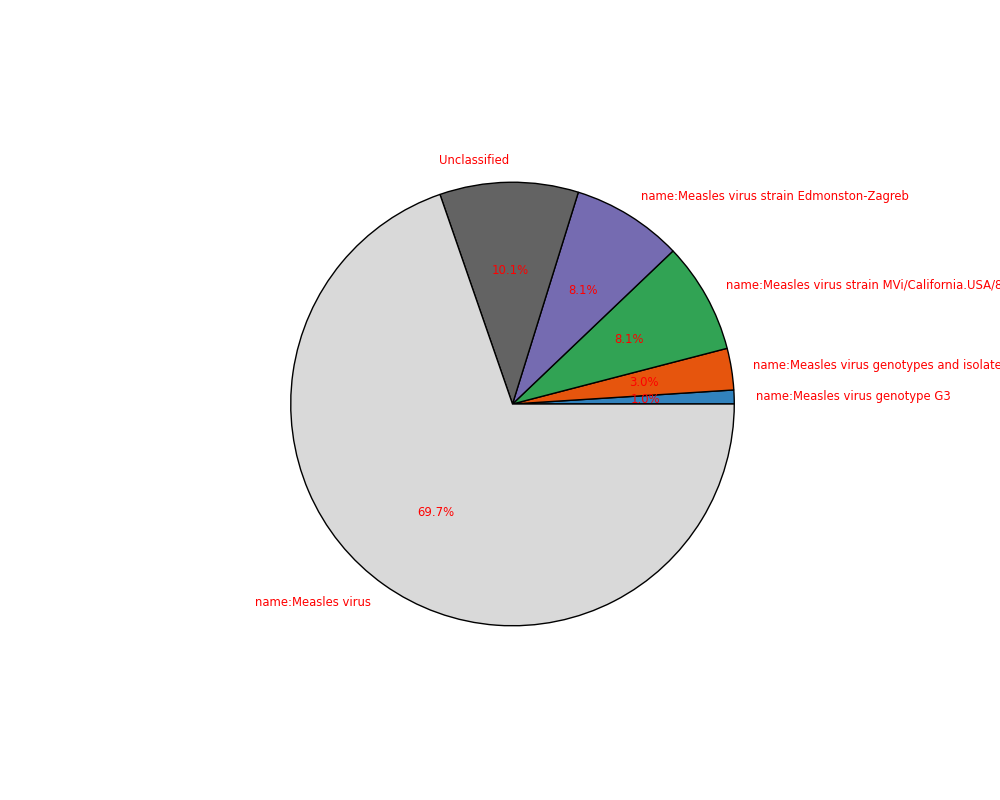
sequana_taxonomy performs a quick taxonomy of your FastQ. This requires dedicated databases to be downloaded.
Sequanix: GUI for snakemake workflows, a GUI for Snakemake workflows (hence Sequana pipelines as well)
The sequana pipelines are various. Since March 2020, they have their own independent life within dedicated github repositories. You may find pipelines for NGS quality control (e.g. adapters removal, phix removal, trimming of bad quality bases), variant calling, characterisation of the genome coverage, taxonomic classification, de-novo assembly, Variant calling, RNA-seq, etc. See the Pipelines section for more information.
Sequana can be used by developers to create new pipelines and by users in the form of applications ready for production. Moreover, Sequanix can be used to set the parameters of pipelines and execute them easily with a graphical user interface.
To join the project, please let us know on github.
Installation
conda install sequana
Examples
Visit our example gallery to use the Python library
NGS pipelines
Learn about available Snakemake pipelines
Standalone applications
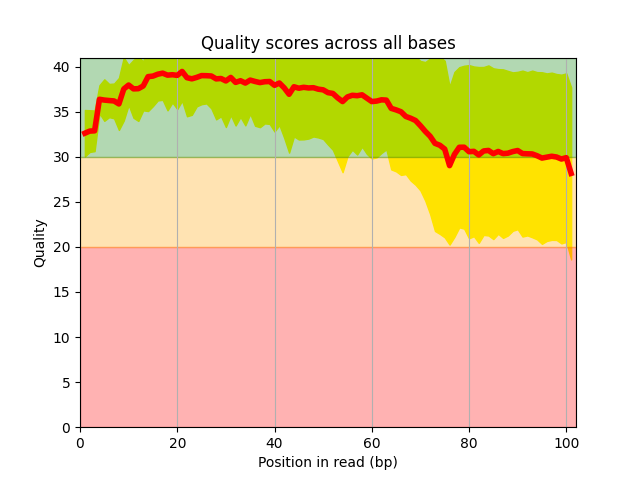
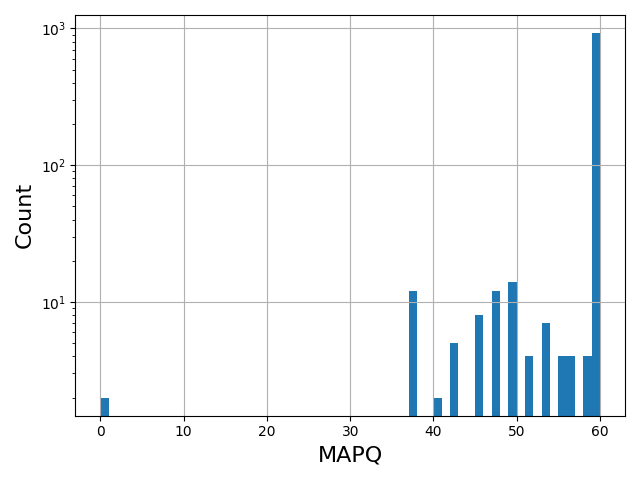
Standalone applications including Sequanix (GUI for snakemake) and the sequana_coverage tool.
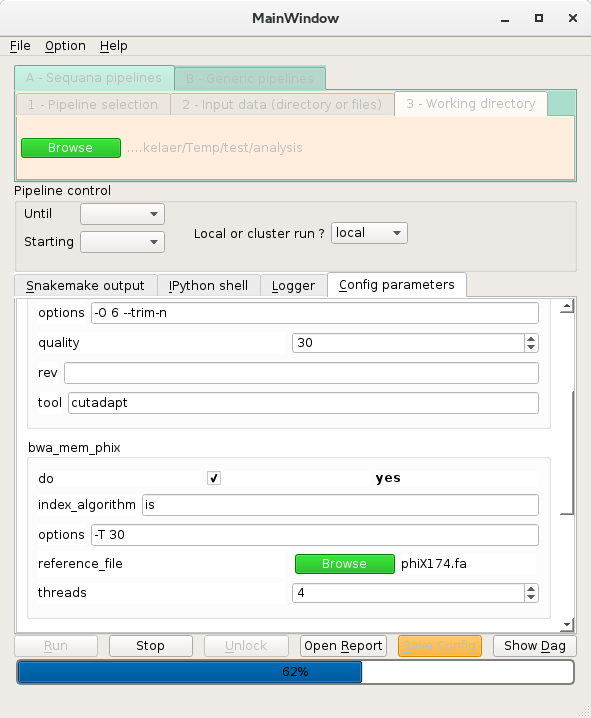
User guide and reference
- 1. Installation
- 2. Overview
- 3. Tutorial
- 4. Pipelines
- 5. Gallery
- 6. Case Examples
- 7. Applications (standalone)
- 8. Sequanix Tutorial
- 8.1. Quick Installation
- 8.2. Introduction
- 8.3. Sequana pipeline: the quality control example
- 8.4. Generic pipeline: a minimalist example with no configuration file
- 8.5. Generic pipeline: a minimalist example with a configuration file
- 8.6. Dialogs and running analysis locally or on a cluster
- 8.7. FAQS
- 9. Developer guide
- 10. Wrappers
- 10.1. bowtie2/align
- 10.2. bowtie2/build
- 10.3. add_read_group
- 10.4. bam_coverage
- 10.5. bcl2fastq
- 10.6. blast
- 10.7. busco
- 10.8. bz2_to_gz
- 10.9. canu
- 10.10. consensus
- 10.11. digital_normalisation
- 10.12. dsrc_to_gz
- 10.13. falco
- 10.14. fastp
- 10.15. fastq_stats
- 10.16. fastqc
- 10.17. feature_counts
- 10.18. freebayes
- 10.19. freebayes_vcf_filter
- 10.20. gz_to_bz2
- 10.21. hmmbuild
- 10.22. hmmscan
- 10.23. index
- 10.24. longorfs
- 10.25. macs3
- 10.26. makeblastdb
- 10.27. mark_duplicates
- 10.28. minimap2
- 10.29. multiqc
- 10.30. polypolish
- 10.31. predict
- 10.32. prokka
- 10.33. quast
- 10.34. rulegraph
- 10.35. sambamba_filter
- 10.36. sambamba_markdup
- 10.37. samtools_depth
- 10.38. sequana_coverage
- 10.39. sequana_taxonomy
- 10.40. snpeff
- 10.41. snpeff_add_locus_in_fasta
- 10.42. sort
- 10.43. spades
- 10.44. trinity
- 10.45. trinity_quantify
- 10.46. unicycler
- 11. References
- 11.1. Assembly and contigs related
- 11.2. BAMTOOLS related
- 11.3. Coverage (bedtools module)
- 11.4. CIGAR tools
- 11.5. Coverage (theoretical)
- 11.6. Access to online database (e.g. ENA)
- 11.7. Enrichment
- 11.8. Experimental design
- 11.9. FASTQ module
- 11.10. FASTA module
- 11.11. Feature counts module
- 11.12. Sequence and genomic modules
- 11.13. Kmer module
- 11.14. Taxonomy related (Kraken - Krona)
- 11.15. Pacbio module
- 11.16. Phred quality
- 11.17. RiboDesigner
- 11.18. RNAdiff
- 11.19. Running median
- 11.20. Snpeff module
- 11.21. General tools
- 11.22. Peaks
- 11.23. Format IO
- 11.24. VCF module
- 11.25. Module Reports
- 11.26. Wrapper to other tools
- 11.27. Misc
- 12. References (enrichment)
- 13. References (stats)
- 14. References (Viz)
- 15. FAQS
- 15.1. Conda related
- 15.2. What are the dependencies
- 15.3. Installation issues
- 15.4. Expected input format
- 15.5. Sequanix related
- 15.6. QXcbConnection issue
- 15.7. Variant Calling pipeline
- 15.8. Quality Control pipeline
- 15.9. Singularity
- 15.10. I got a error "main thread is not in the main loop"
- 15.11. Installation issue on Mac
- 15.12. pbindex
- 16. Glossary