Applications (standalone)#
Sequanix: GUI for snakemake workflows#
- Overview:
a Graphical User Interface (GUI) for Sequana pipelines and any Snakemake-based workflows.
- Status:
Production
- Name:
sequanix
This GUI can be used to load Snakefile and their configuration file. A working directory has to be set. Once done, the configuration file can be changed in the GUI. Finally, one can run the snakefile and see the progress. Tooltips are automatically created from the configuration file (if documented).
Since snakemake has the ability to run jobs locally or on a cluster, this application can also be run either locally or a distributed computing platform (e.g., cluster with slurm scheduler). Of course, this means you can use a X environment on your cluster (ssh -X should do it).
Just type sequanix in a shell.
Note
tested under Linux only. However, Mac and Windows users should be able to use it since it is based on Python and PyQt. Again, we strongly advice to use Anaconda to install all required dependencies
Here is a snapshot.
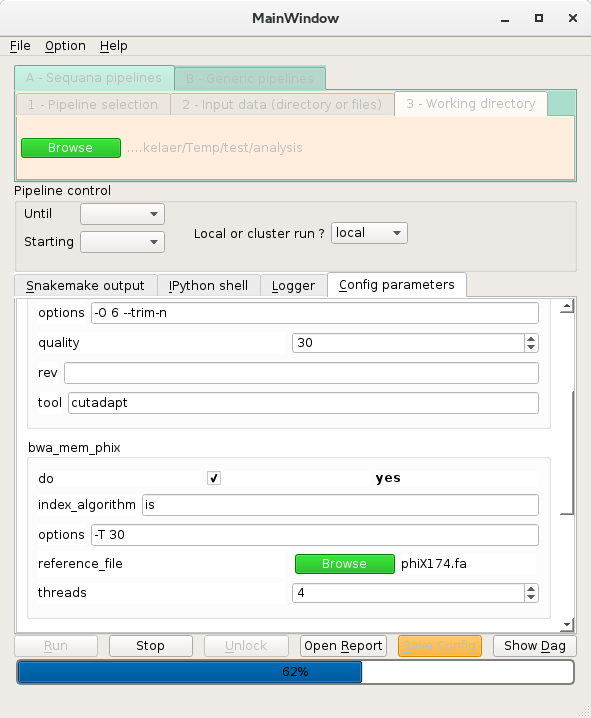
See also
see Sequanix Tutorial for details
sequana_coverage#
- Description:
Show coverage and interval of confidence to identify under and over represented genomic regions.
- Status:
Production
- Help:
please use sequana_coverage
--help- Sequana:
See
GenomeCovto use the coverage in your own script.- Gallery:
See examples in the gallery
Starting from a BED file and its reference, one can use this command in a shell:
sequana_coverage --input JB409847.sorted.bed -o
--reference JB409847.fa --show-html
It creates an HTML report with various images showing the coverage and GC versus coverage plots. It also provides a set of CSV files with low or high coverage regions (as compared to the average coverage).
See also
the underlying algorithm is described in details in the documentation
(sequana.bedtools.GenomeCov).
sequana_summary#
- Description:
Prints basic statistics about a set of NGS input files. Currently handles Fastq (gzipped or not) or BED files (coverage).
- Usage:
sequana summary file1.fastq.gz
sequana_mapping#
- Description:
a simple application to map reads onto a genome given one or two FastQ files (gzipped) and a reference.
sequana_mapping --file1 H1_R1.fastq.gz --file2 H1_R2.fastq.gz --reference temp.fa
will map all reads on the reference using bwa.
sequana_taxonomy#
- Description:
Creates a HTML document with Krona and pie chart of taxonomic content of a FastQ file (paired or not). Uses Kraken, Krona and a dedicated Sequana database.
- Help:
sequana_taxonomy --help- Status:
Production
- Sequana:
see
sequana.kraken- Gallery:
You will need to download databases. We provide a toy example:
sequana_taxonomy --download toydb