RNA-seq#
current version:?
This is is the rnaseq pipeline from the Sequana project
Installation
You must install Sequana first:
pip install sequana
Then, just install this package:
pip install sequana_rnaseq
For all dependencies (see hereafter), you can use conda. Another experimental solution is to use damona:
pip install damona damona install sequana_tools
This will install all required dependencies.
Usage
sequana_pipelines_rnaseq --help sequana_pipelines_rnaseq --input-directory DATAPATH --genome-directory genome --aligner star
This creates a directory with the pipeline and configuration file. You will then need to execute the pipeline:
cd rnaseq sh rnaseq.sh # for a local run
This launch a snakemake pipeline. If you are familiar with snakemake, you can retrieve the pipeline itself and its configuration files and then execute the pipeline yourself with specific parameters:
snakemake -s rnaseq.rules -c config.yaml --cores 4 --stats stats.txt
Or use sequanix interface.
Requirements
This pipelines requires lots of third-party executable(s). Here is a list that may change. A Message will inform you would you be missing an executable:
- bowtie
- bowtie2
- STAR
- featureCounts (subread package)
- picard
- multiqc
You can install most of the tools using damona:
damona create --name sequana_tools damona activate sequana_tools damona install sequana_tools
Or use the conda.yaml file available in this repository. If you start a new environment from scratch, those commands will create the environment and install all dependencies for you:
conda create --name sequana_env python 3.7.3 conda activate sequana_env conda install -c anaconda qt pyqt>5 pip install sequana pip install sequana_rnaseq conda install --file https://raw.githubusercontent.com/sequana/rnaseq/master/conda.yaml
Details
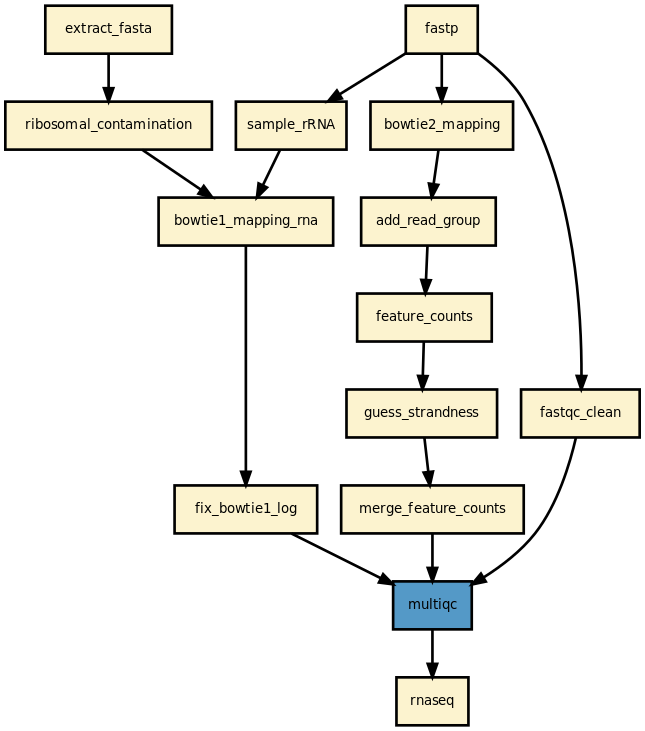
This pipeline runs a RNA-seq analysis of sequencing data. It runs in parallel on a set of input FastQ files (paired or not). A brief HTML report is produced together with a MultiQC report.
This pipeline is complex and requires some expertise for the interpretation. Many online-resources are available and should help you deciphering the output.
Yet, it should be quite straigtforward to execute it as shown above. The pipeline uses bowtie1 to look for ribosomal contamination (rRNA). Then, it cleans the data with cutapdat if you say so (your data may already be pre-processed). If no adapters are provided (default), reads are trimmed for low quality bases only. Then, mapping is performed with standard mappers such as star or bowtie2 (--aligner option). Finally, feature counts are extracted from the previously generated BAM files. We guess the strand and save the feature counts into the directoy ./rnadiff/feature_counts.
The pipelines stops there. However, RNA-seq analysis are followed by a different analysis (DGE hereafter). Although the DGE is not part of the pipeline, you can performed it with standard tools using the data in ./rnadiff directory. One such tool is provided within our framework (based on the well known DEseq2 software).
Using our framework:
cd rnadiff
sequana rnadiff --design design.csv --features all_features.out --annotation ANNOT \
--feature-name FEAT --attribute-name ATTR
where ANNOT is the annotation file of your analysis, FEAT and ATTR the attribute and feature used in your analysis (coming from the annotation file).
This produces a HTML repot summarizing you differential analysis.
Rules and configuration details
Here is the latest documented configuration file to be used with the pipeline. Each rule used in the pipeline may have a section in the configuration file.
Warning
the RNAseQC rule is switch off and is not currently functional in version 0.9.X
Changelog
| Version | Description |
|---|---|
| 0.15.2 |
|
| 0.15.1 |
|
| 0.15.0 |
|
| 0.14.2 |
|
| 0.14.1 |
|
| 0.14.0 |
|
| 0.13.0 |
|
| 0.12.1 |
|
| 0.12.0 |
|
| 0.11.0 |
|
| 0.10.0 |
|
| 0.9.20 |
|
| 0.9.19 |
|
| 0.9.18 |
|
| 0.9.17 |
|
| 0.9.16 |
|
| 0.9.15 |
|
| 0.9.14 |
|
| 0.9.13 |
|
| 0.9.12 |
|
| 0.9.11 |
|
| 0.9.10 |
|
| 0.9.9 |
|
| 0.9.8 |
|
| 0.9.7 |
|
| 0.9.6 |
|
| 0.9.5 |
|
| 0.9.4 | |
| 0.9.3 | if a fastq_screen.conf is provided, we switch the fastqc_screen section ON automatically |
| 0.9.0 | Major refactorisation.
|