Library user guide#
Use this page if you intend to call Sequana from Python or a Jupyter notebook — to read NGS file formats, compute metrics, or assemble report sections from a script.
For end-users running pre-built pipelines, see Pipeline user guide instead.
Test data#
Sequana ships a small data folder accessible from Python via
sequana_data():
from sequana import sequana_data
filename = sequana_data('JB409847.bed')
A complete list of bundled files is in sequana.datatools.
Coverage from a BED file#
Read a BED file produced by bedtools genomecov:
from sequana import SequanaCoverage
gc = SequanaCoverage(filename)
Select a chromosome, compute the running median and z-score:
chrom = gc[0]
chrom.running_median(n=5001, circular=True)
chrom.compute_zscore()
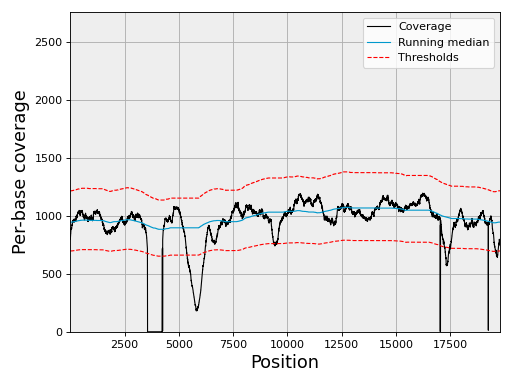
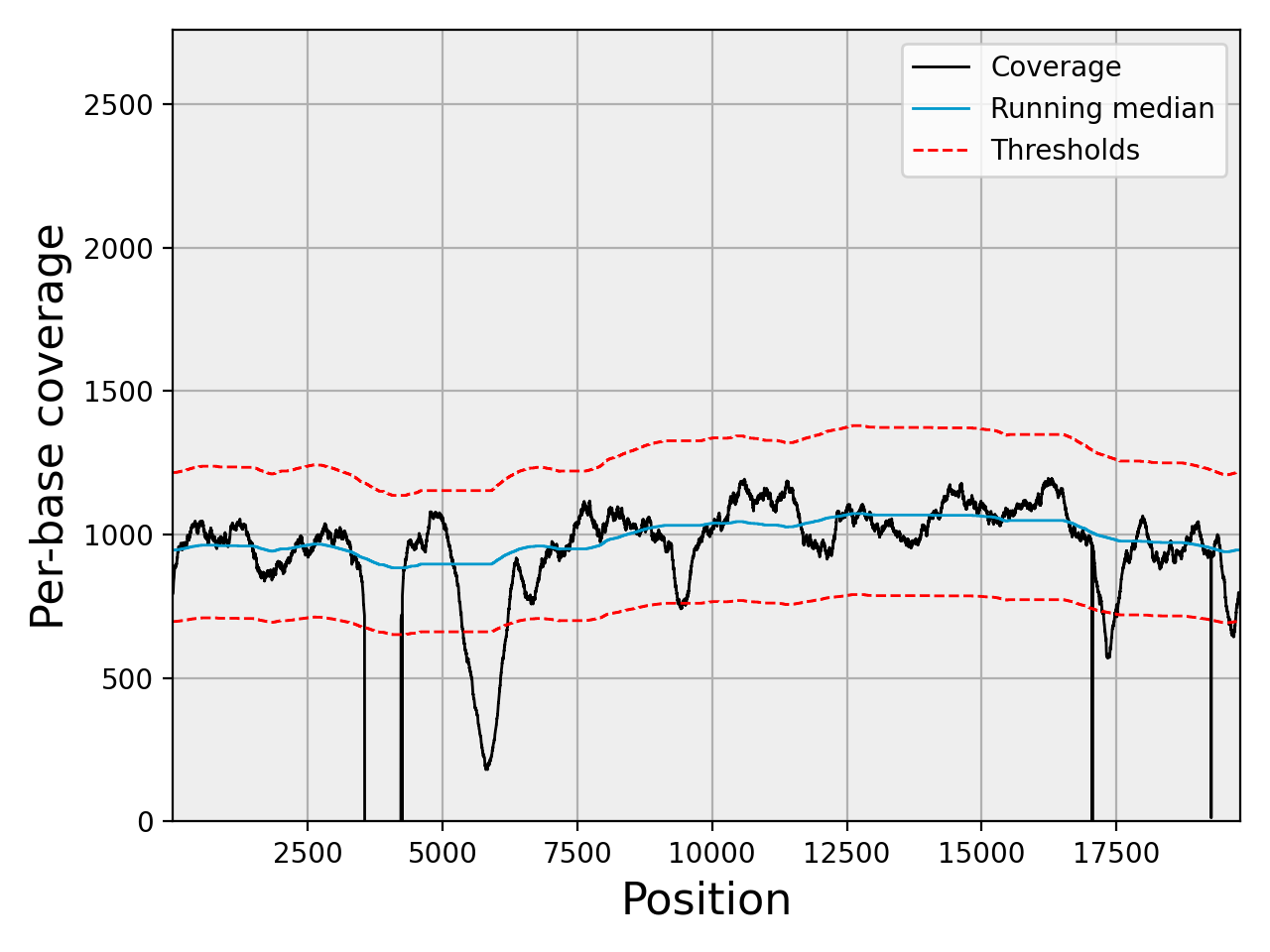
Plot the coverage with its 3-sigma confidence band:
chrom.plot_coverage()
(Source code, png, hires.png, pdf)
{kind=link}
{kind=link}
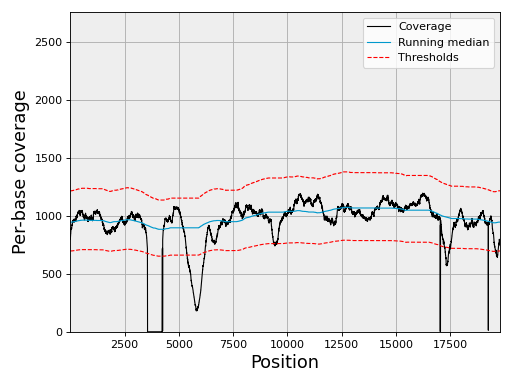
A matching notebook is at notebooks/coverage.ipynb.
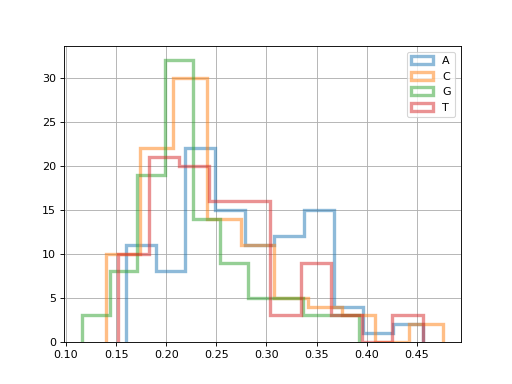
FastQ inspection#
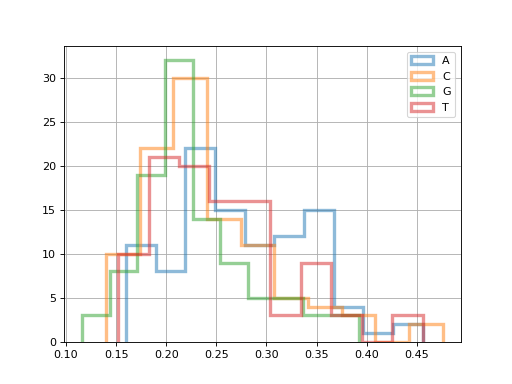
The FastQC class exposes per-read metrics:
from sequana import FastQC, sequana_data
fastqc = FastQC(sequana_data("test.fastq"))
print(fastqc.fastq)
for x in 'ACGT':
fastqc.get_actg_content()[x].hist(
alpha=0.5, label=x, histtype='step', lw=3, bins=10)
from sequana import FastQC, sequana_data
fastqc = FastQC(sequana_data("test.fastq"))
for x in 'ACGT':
fastqc.get_actg_content()[x].hist(
alpha=0.5, label=x, histtype='step', lw=3, bins=10)
from pylab import legend
legend()
(Source code, png, hires.png, pdf)
{kind=link}
{kind=link}
Building HTML report sections from Python#
Sequana's pipeline reports are themselves assembled from reusable building
blocks in sequana.modules_report. You can call them on your own data.
Example for a BAM file:
from sequana import BAM, sequana_data
from sequana.modules_report.bamqc import BAMQCModule
BAMQCModule(sequana_data("test.bam", "testing"), "bam.html")
The generated bam.html is a self-contained page (see
bam.html for a rendered example).
To build a brand-new report module, see Module reports in the developer guide.
Where to look next#
API reference — the full module index.
Gallery — Sphinx-Gallery of short, runnable scripts.
Notebooks — Jupyter notebooks demonstrating BAM, coverage, FastQ, feature counts and ribodesigner workflows.
CLI reference — the
sequanaCLI sub-commands (some of these wrap the Python API).