References (IO / file formats)#
FASTA#
Utilities to manipulate FastA files
- class FastA(filename, verbose=False)[source]#
Class to handle FastA files
Works like an iterator:
from sequana import FastA f = FastA("test.fa") read = next(f)
names and sequences can be accessed with attributes:
f.names f.sequences
- GC_content_per_sequence(exclude=[])[source]#
Return a dictionary of GC content (in percent) for each contig/chromosome.
N (and any non-ACGT character) are excluded from the total so that the GC content is computed as G+C over A+C+G+T only.
- Parameters:
exclude (list) -- list of contig/chromosome names to skip.
- Returns:
dict mapping each sequence name to its GC content in percentage.
- GC_content_sequence(sequence)[source]#
Return GC content in percentage of a sequence (N excluded from the total)
Deprecated since version Use:
GC_content_per_sequence()instead.
- property comments#
- explode(outdir='.')[source]#
extract sequences from one fasta file and save them into individual files
- filter(output_filename, names_to_keep=None, names_to_exclude=None)[source]#
save FastA excluding or including specific sequences
- find_gaps()[source]#
Identify NNNNs in data
returns a dictionary. keys are the chromosomes' names values is a list. the first item is the number of Ns. the next items are the gaps' positions
- format_contigs_denovo(output_file, len_min=500)[source]#
Remove contigs with sequence length below specific threshold.
Example:
from sequana import FastA contigs = FastA("denovo_assembly.fasta") contigs.format_contigs_denovo("output.fasta", len_min=500)
- get_cumulative_sum(mode='mixed', exclude=[])[source]#
Compute the cumulative sum of values from a dictionary, sorted by name.
This method returns two lists:
A list of names sorted according to the specified mode.
A list of cumulative sums corresponding to the lengths of the sorted names.
Sorting behavior:
If
mode="mixed"(default), names containing numbers are sorted naturally, meaning numerical values are sorted as integers while preserving non-numeric strings.If
mode="alphanum", all names are treated as strings and sorted lexicographically.
- Parameters:
- Returns:
tuple
(sorted_names, cumulative_sums)wheresorted_namesis the list of names sorted by the selected mode andcumulative_sumsis the cumulative sum of corresponding values.
Example:
If input is
['1', 'maxi', '10', '2'], mixed mode returns['1', '2', '10', 'maxi'], ensuring['1', 10, 2, 'maxi']is correctly ordered as[1, 2, '10', 'maxi'].
- get_stats()[source]#
Return a dictionary with basic statistics
N the number of contigs, the N50 and L50, the min/max/mean contig lengths and total length.
- property lengths#
- property names#
- plot_GC_content(sorting='mixed', max_contigs=50, kind='line', hline=True, rotation=90, hold=False, exclude=[])[source]#
Plot the GC content (in percent) per contig/chromosome.
The x-axis is the contig/chromosome name and the y-axis is the GC content.
- Parameters:
sorting (str) --
"mixed"(natural sort) or"alphanum"(lexicographic) ordering of the names along the x-axis.max_contigs (int) -- keep only the
max_contigslongest contigs to keep the plot readable. Set to-1to plot all of them.kind (str) --
"line"or"bar".hline (bool) -- draw a horizontal dashed line at the genome-wide GC content.
rotation (int) -- rotation of the x-axis tick labels.
hold (bool) -- if False (default), clear the figure before plotting.
exclude (list) -- list of contig/chromosome names to skip.
- save_collapsed_fasta(outfile, ctgname, width=80, comment=None)[source]#
Concatenate all contigs and save results
- select_random_reads(N=None, output_filename='random.fasta')[source]#
Select random reads and save in a file
- property sequences#
- property sorted_mixed_names#
- property sorted_names#
- summary(max_contigs=-1)[source]#
returns summary and print information on the stdout
This method is used when calling sequana standalone
sequana summary test.fasta
- class Protein(sequence, use_exotic_amino_acid=True)[source]#
Simple Protein class
>>> d = Protein("MALWMRLLPLLALLALWGPD") >>> d.check() >>> d.stats()
Constructor
A sequence is just a string stored in the
sequenceattribute. It has properties related to the type of alphabet authorised.- Parameters:
sequence (str) -- May be a string of a Fasta File, in which case only the first sequence is used.
complement_in
complement_out
letters -- authorise letters. Used in
check()only.
Todo
use counter only once as a property
FASTQ#
Utilities to manipulate FASTQ and Reads
- class FastQ(filename, verbose=False)[source]#
Class to handle FastQ files
Some of the methods are based on pysam but a few are also original to sequana. In general, input can be zipped ot not and output can be zipped or not (based on the extension).
An example is the
extract_head()method:f = FastQ("input_file.fastq.gz") f.extract_head(100000, output='test.fastq') f.extract_head(100000, output='test.fastq.gz')
equivalent to:
zcat myreads.fastq.gz | head -100000 | gzip > test100k.fastq.gz
An efficient implementation to count the number of lines is also available:
f.count_lines()
or reads (assuming 4 lines per read):
f.count_reads()
Operators available:
equality ==
- extract_head(N, output_filename)[source]#
Extract the heads of a FastQ files
- Parameters:
This function is convenient since it takes into account the input file being compressed or not and the output file being compressed ot not. It is in general 2-3 times faster than the equivalent unix commands combined together but is 10 times slower for the case on uncompressed input and uncompressed output.
Warning
this function extract the N first lines and does not check if there are empty lines in your FastQ/FastA files.
- filter(identifiers_list=[], min_bp=None, max_bp=None, progress=True, output_filename='filtered.fastq')[source]#
Save reads in a new file if there are not in the identifier_list
- joining(pattern, output_filename)[source]#
not implemented
zcat Block*.fastq.gz | gzip > combined.fastq.gz
- property n_lines#
return number of lines (should be 4 times number of reads)
- property n_reads#
return number of reads
- plot_GC_read_len(hold=False, fontsize=12, bins=[200, 60], grid=True, cmap='BrBG', maxreads=10000)[source]#
- rewind()[source]#
Allows to iter from the beginning without openning the file or creating a new instance.
- select_random_reads(N=None, output_filename='random.fastq', progress=True)[source]#
Select random reads and save in a file
- Parameters:
If you have a pair of files, the same reads must be selected in R1 and R2.:
f1 = FastQ(file1) selection = f1.select_random_reads(N=1000) f2 = FastQ(file2) f2.select_random_reads(selection)
Changed in version 0.9.8: use list instead of set to keep integrity of paired-data
- select_reads(read_identifiers, output_filename=None, progress=True)[source]#
identifiers must be the name of the read without starting @ sign and without comments.
- to_fasta(output_filename='test.fasta')[source]#
Slow but works for now in pure python with input compressed data.
- class FastQC(filename, max_sample=500000, verbose=True, skip_nrows=0)[source]#
Simple QC diagnostic
Similarly to some of the plots of FastQC tools, we scan the FastQ and generates some diagnostic plots. The interest is that we'll be able to create more advanced plots later on.
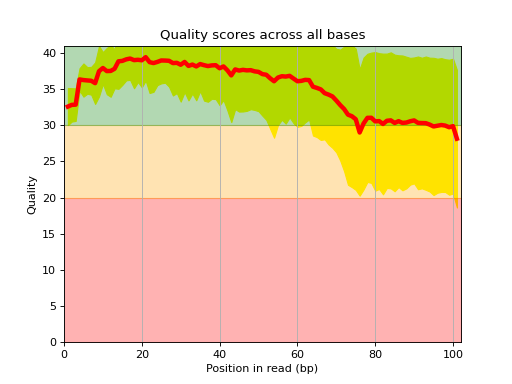
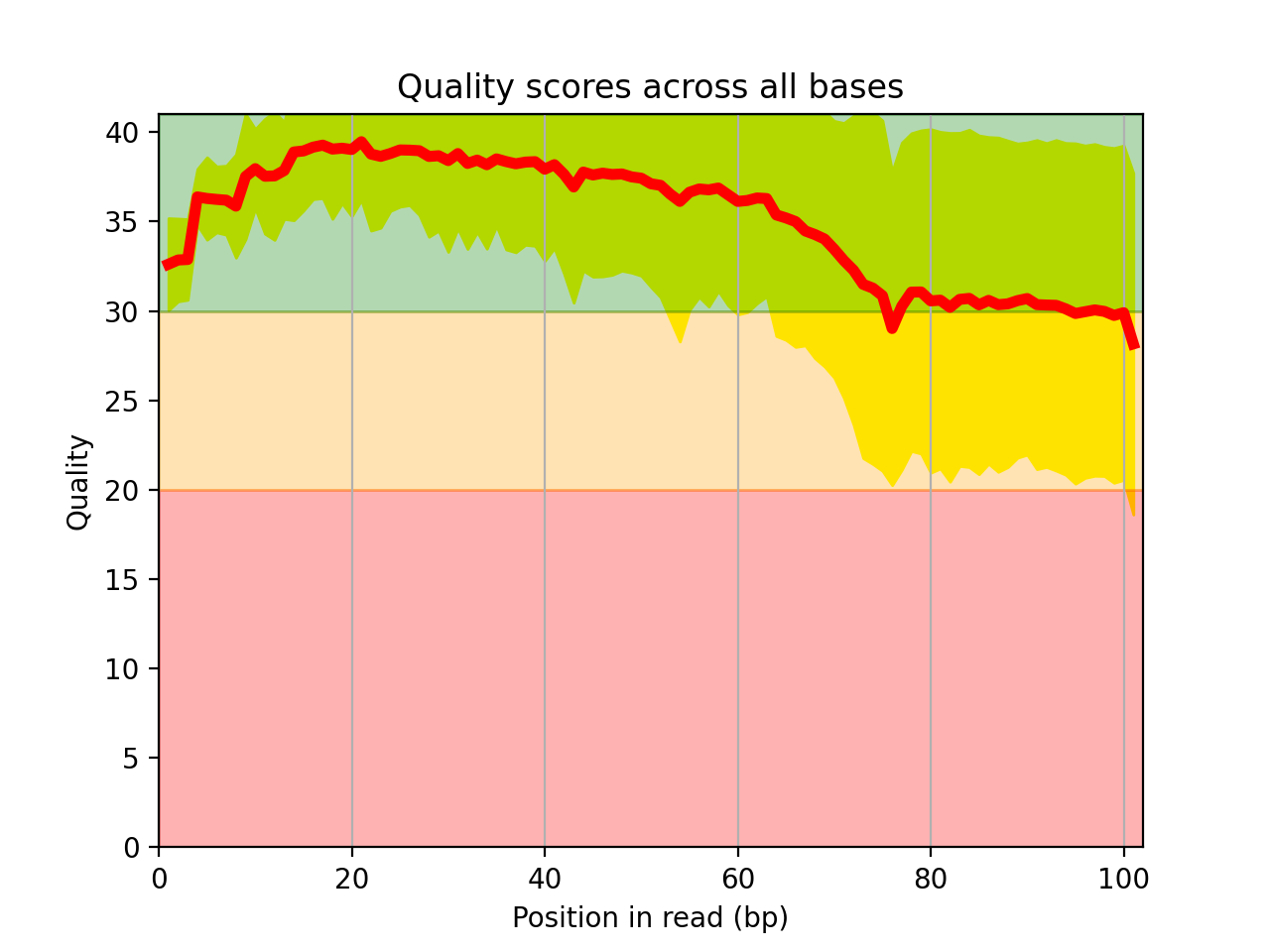
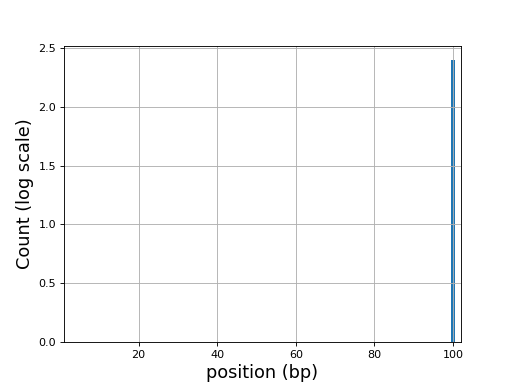
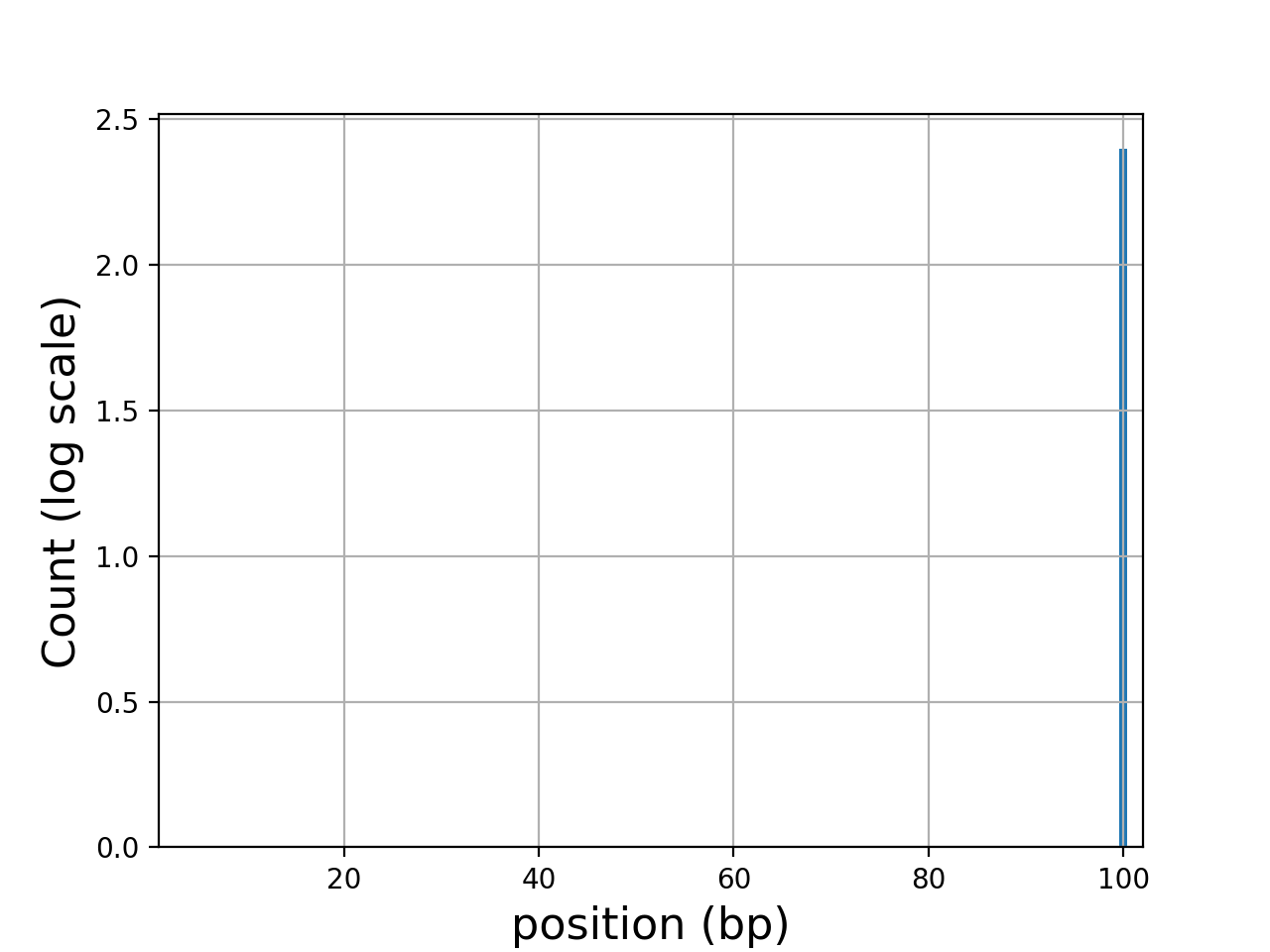
Here is an example of the boxplot quality across all bases:
from sequana import sequana_data from sequana import FastQC filename = sequana_data("test.fastq", "doc") qc = FastQC(filename) qc.boxplot_quality()
(
Source code,png,hires.png,pdf)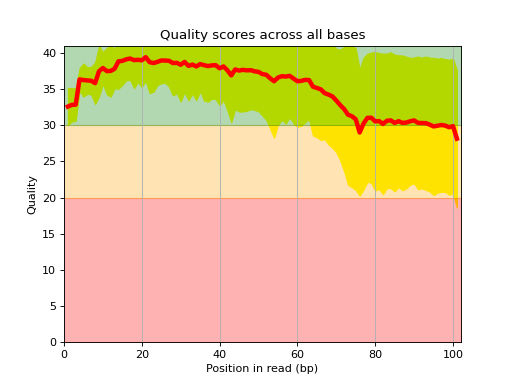
Warning
some plots will work for Illumina reads only right now
Note
Although all reads are parsed (e.g. to count the number of nucleotides, some information uses a limited number of reads (e.g. qualities), which is set to 500,000 by deafult.
constructor
- Parameters:
filename
max_sample (int) -- Large files will not fit in memory. We therefore restrict the numbers of reads to be used for some of the statistics to 500,000. This also reduces the amount of time required to get a good feeling of the data quality. The entire input file is parsed tough. This is required for instance to get the number of nucleotides.
- boxplot_quality(hold=False, ax=None)[source]#
Boxplot quality
Same plots as in FastQC that is average quality for all bases. In addition a 1 sigma error enveloppe is shown (yellow).
Background separate zone of good, average and bad quality (arbitrary).
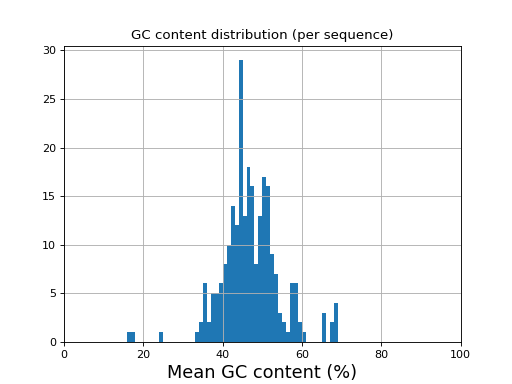
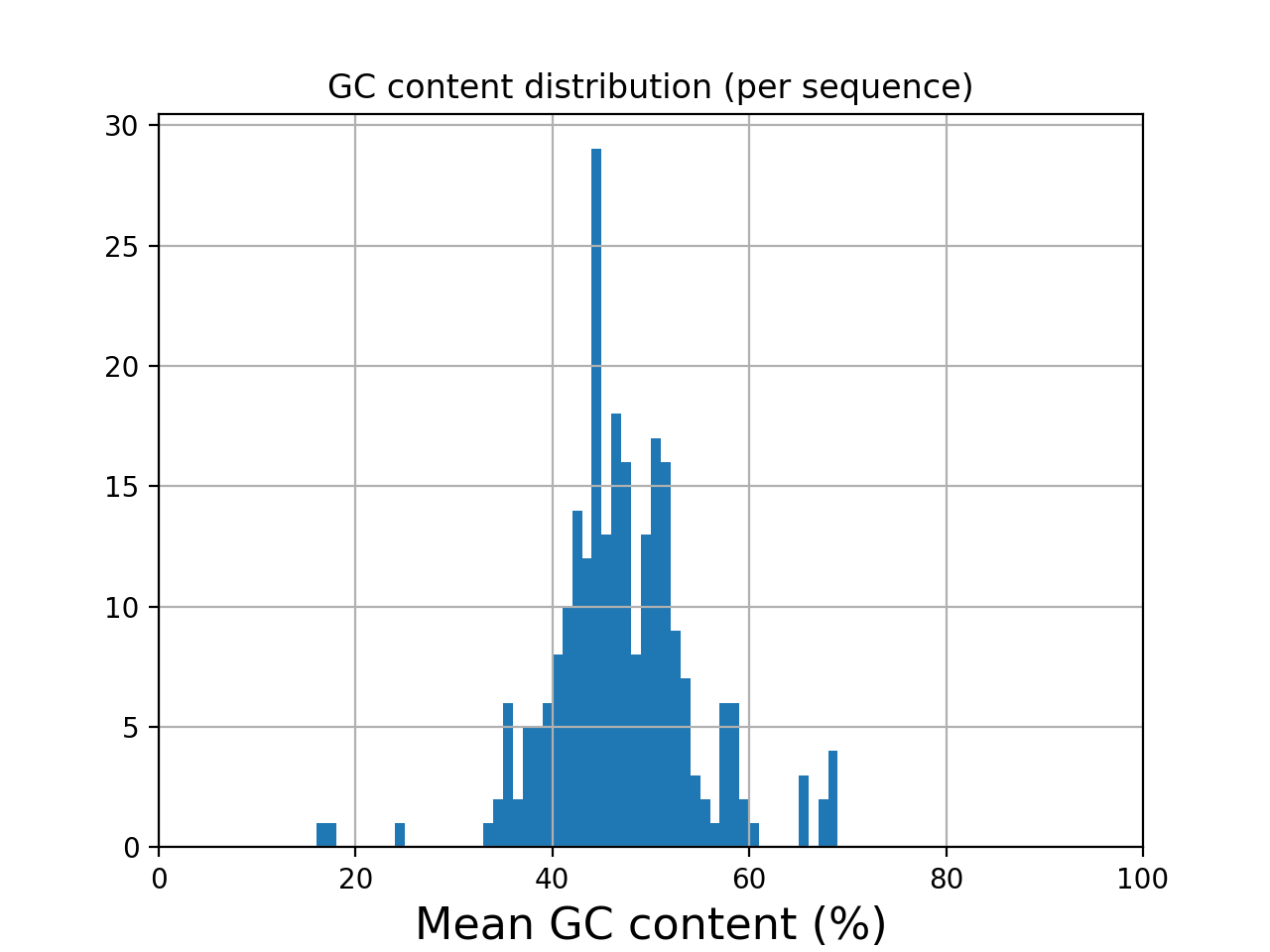
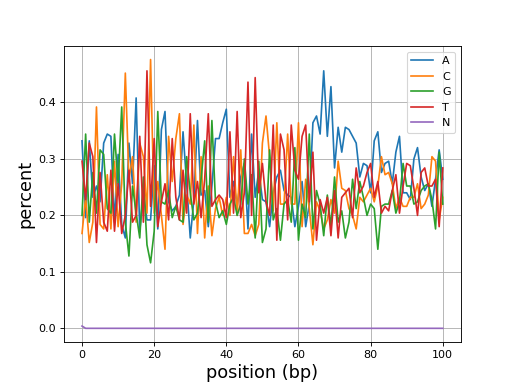
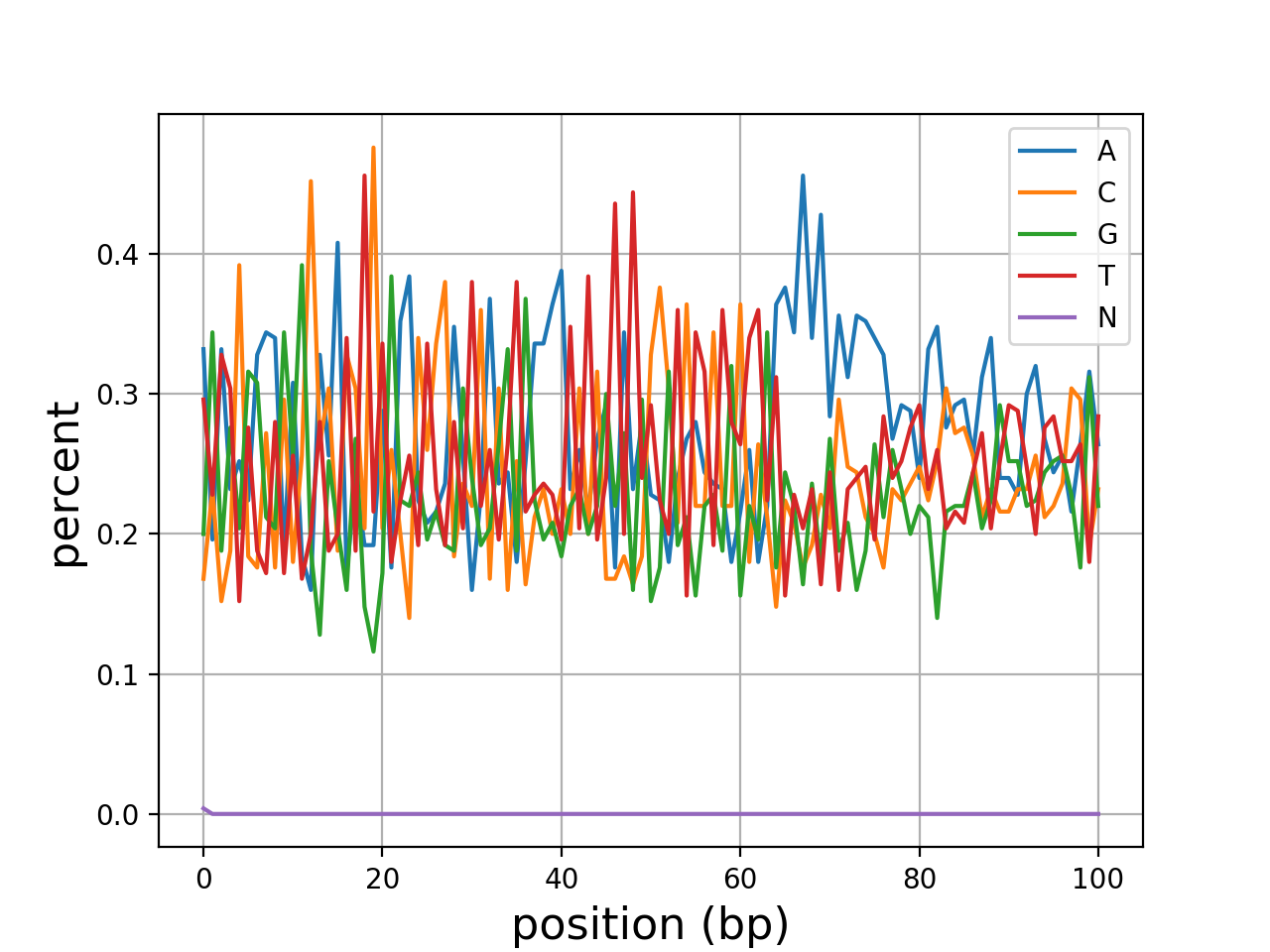
- histogram_gc_content()[source]#
Plot histogram of GC content
from sequana import sequana_data from sequana import FastQC filename = sequana_data("test.fastq", "doc") qc = FastQC(filename) qc.histogram_gc_content()
(
Source code,png,hires.png,pdf)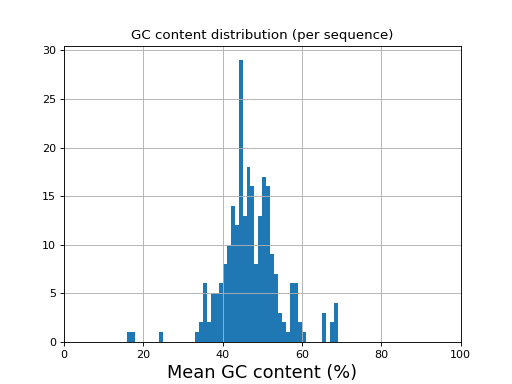
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
- class Identifier(identifier, version='unknown')[source]#
Class to interpret Read's identifier
Warning
Implemented for Illumina 1.8+ and 1.4 . Other cases will simply stored the identifier without interpretation
>>> from sequana import Identifier >>> ident = Identifier('@EAS139:136:FC706VJ:2:2104:15343:197393 1:Y:18:ATCACG') >>> ident.info['x_coordinate'] '2'
Currently, the following identifiers will be recognised automatically:
- Illumina_1_4:
An example is
@HWUSI-EAS100R:6:73:941:1973#0/1
- Illumina_1_8:
An example is:
@EAS139:136:FC706VJ:2:2104:15343:197393 1:Y:18:ATCACG
Other that could be implemented are NCBI
@FSRRS4401BE7HA [length=395] [gc=36.46] [flows=800] [phred_min=0] [phred_max=40] [trimmed_length=95]
Information can also be found here http://support.illumina.com/help/SequencingAnalysisWorkflow/Content/Vault/Informatics/Sequencing_Analysis/CASAVA/swSEQ_mCA_FASTQFiles.htm
- class FastqSplitter(filename: str | Path)[source]#
- by_part(n_parts: int, pattern: str, gzip_output: bool = False, buffer_size: int = 10000)[source]#
Split FASTQ into N parts with approximate read counting.
This is a MultiQC plugin re-used temporarely here
BED / wig / mpileup#
- class BED(filename)[source]#
a structure to read and manipulate BED files (12-column file)
columns are defined as chromosome name, start and end, gene_name, score, strand, CDS start and end, blcok count, block sizes, block starts:
- get_exons()[source]#
Extract exon regions from input BED file.
Uses the first (chromosome name), second (chromosome start), 11th and 12th columns (exon start and size) of a 12-columns BED file.
from sequana import sequana_data from sequana import BED b = BED(sequana_data("hg38_chr18.bed")) b.get_exons()
- yield_bigwig_by_chromosome(filename)[source]#
Yield
(chrom, entries)from a binary BigWig file.for chrom, entries in yield_bigwig_by_chromosome("coverage.bw"): print(f"Chromosome: {chrom}, {len(entries)} entries") print(entries[:3])
Each
entriesis a list of(start, value)tuples, one per interval stored in the BigWig (e.g. one perbamCoveragebin).pyBigWigis imported lazily.
Tools to manipulate output of mpileup software
GFF / GTF / GenBank / GFA#
- class GFF3(filename, skip_types=['biological_region'], light=False)[source]#
Read a GFF file, version 3
See also
g = GFF3(filename) # first call is slow g.df # print info about the different feature types g.features # prints info about duplicated attributes: g.get_duplicated_attributes_per_genetic_type(self)
On eukaryotes, the reading and processing of the GFF may take a while. On prokaryotes, it should be pretty fast (a few seconds). To speed up the eukaryotes case, we skip the processing biological_regions (50% of the data in mouse).
You can do a lot with this class because your GFF is stored as a dataframe and and therefore be easily processed.
Sometimes, CDS are missing:
g = GFF3() g.add_CDS_and_mRNA("test.gff")
Initialise a GFF3 reader.
- Parameters:
- Raises:
IOError -- if filename does not exist.
- add_CDS_and_mRNA(output, gene_ID='gene_id')[source]#
Rewrite the GFF file adding synthetic mRNA and CDS entries for every gene.
Some annotation files (e.g. for bacteria or simple eukaryotes) contain only
genefeatures without the correspondingmRNA/CDSchildren required by certain downstream tools. This method reads the source GFF line by line and, for everygenefeature, emits the original gene line followed by a syntheticmRNAline and a syntheticCDSline sharing the same coordinates.
- add_directon_index()[source]#
Assign each gene its positional index within its directon.
For each gene, sets its ordinal position inside the directon it belongs to (assuming the GFF is sorted by chromosome and start coordinate).
Note
Not yet implemented; this method is a stub reserved for a future feature.
- add_intergenic_regions()[source]#
Append intergenic regions to the internal dataframe.
Calls
get_intergenic_regions()and concatenates the result with the existing annotation dataframe. Subsequent calls are no-ops (the operation is only performed once).
- add_regions_and_save_gff(filename)[source]#
add missing region from the sequence-region found in the header.
- clean_gff_line_special_characters(text)[source]#
Simple leaner of gff lines that may contain special characters
- property contig_names#
- property df#
Build and cache a
pandas.DataFramefrom the GFF3 file.The dataframe contains one row per GFF3 record. Column names match the nine standard GFF3 fields (
seqid,source,genetic_type,start,stop,score,strand,phase,attributes) plus one column per attribute key found in the file.The result is cached so subsequent accesses are instantaneous.
- directon_to_bed(output='directons.bed', colors={'+': '255,0,0', '-': '0,0,255'})[source]#
Write directon information to a BED9 file.
Each directon is written as one line in the output BED file with colour-coding by strand (red for
+, blue for-by default).
- property directons#
Compute directons: groups of consecutive genes sharing the same strand.
A directon is a maximal set of adjacent genes transcribed from the same strand. The method groups the annotation dataframe by chromosome, detects strand changes to assign each gene to a directon, then aggregates per-directon statistics (start, stop, number of genes).
The result is cached; subsequent calls return the cached value.
- Returns:
DataFrame with columns
seqid,strand,directon_id,start,stop,directon_length, anddirecton_name.- Return type:
pandas.DataFrame
- property features#
Extract unique GFF feature types
This is equivalent to awk '{print $3}' | sort | uniq to extract unique GFF types. No sanity check, this is suppose to be fast.
Less than a few seconds for mammals.
- get_PTU()[source]#
Compute Polycistronic Transcription Units (PTUs).
PTUs are contiguous groups of genes transcribed from the same strand. tRNA and rRNA genes are excluded first (they are transcribed by RNA Pol I/III and are not part of the Pol II polycistrons), then directons are computed on the remaining coding genes.
Non-destructive: the object's internal dataframe and cached directons are left untouched.
- Returns:
DataFrame with columns
chromosome,start,stop,strand, andlength(number of genes in the PTU).- Return type:
pandas.DataFrame
- get_duplicated_attributes_per_genetic_type()[source]#
Report duplicated attribute values for each feature type.
Iterates over all feature types found in the GFF file and, for each attribute column, counts how many values appear more than once. The counts are printed to stdout and returned as a
pandas.DataFramewhose columns are feature types and whose rows are attribute names.- Returns:
DataFrame with feature types as columns and attribute names as rows; each cell contains the number of duplicated values.
- Return type:
pandas.DataFrame
- get_intergenic_regions()[source]#
Compute intergenic regions between consecutive genes on each chromosome.
For every chromosome present in the GFF, gaps between consecutive
genefeatures (sorted by start position) are returned as apandas.DataFrame. Each row represents one intergenic region and contains the columnsseqid,start,stop,strand,attributes,source,genetic_type,score, andphase.- Returns:
DataFrame of intergenic regions.
- Return type:
pandas.DataFrame
- get_seqid2size()[source]#
Return a mapping from sequence identifiers to their sizes.
Sizes are read from
regionfeatures in the GFF file (thestopcoordinate of aregionrecord is used as the chromosome/contig size).- Returns:
dict mapping sequence id strings to integer sizes.
- Return type:
- get_simplify_dataframe()[source]#
Method to simplify the gff and keep only the most informative features.
- is_tRNA_or_ribosomal(x)[source]#
Deprecated keep predicate:
Truefor genes to retain.Deprecated since version The: inverted semantics were confusing. Use
_is_tRNA_or_rRNA()(returnsTruefor tRNA/rRNA) instead.
- read_and_save_selected_features(outfile, features=['gene'])[source]#
Stream the GFF file and write only the requested feature types.
Unlike
save_gff_filtered(), this method does not load the whole file into memory; it reads and filters line by line, making it suitable for very large GFF files.
- save_annotation_to_csv(filename='annotations.csv')[source]#
Save the GFF annotation dataframe to a CSV file.
- Parameters:
filename (str) -- path to the output CSV file. Defaults to
"annotations.csv".
- save_as_gff(filename, sortby=['seqid', 'start', 'stop', 'genetic_type'])[source]#
Write the (possibly modified) annotation back to a GFF3 file.
The header comment lines from the original file are preserved. Records are sorted according to sortby using natural sort order so that chromosome identifiers like
chr2sort beforechr10.
- search(pattern)[source]#
Search all columns of the annotation dataframe for a pattern.
Converts every cell to a string and returns all rows where at least one column contains pattern as a substring.
- Parameters:
pattern -- search term (converted to
strinternally).- Returns:
filtered copy of the annotation dataframe.
- Return type:
pandas.DataFrame
- to_bed(output_filename, attribute_name, features=['gene'])[source]#
Experimental export to BED format to be used with rseqc scripts
- Parameters:
attribute_name (str) -- the attribute_name name to be found in the GFF attributes
- to_fasta(ref_fasta, fasta_out, features=['gene'], identifier='ID')[source]#
From a genomic FASTA file ref_fasta, extract regions stored in the gff. Export the corresponding regions to a FASTA file fasta_out.
- Parameters:
ref_fasta -- path to genomic FASTA file to extract rRNA regions from.
fasta_out -- path to FASTA file where rRNA regions will be exported to.
- to_gtf(output_filename='test.gtf', mapper={'ID': '{}_id'})[source]#
Convert the GFF3 file to GTF format (experimental).
Reads the source GFF3 file line by line and rewrites each record in GTF format. Attribute keys are renamed according to mapper which maps GFF3 attribute names to GTF attribute name templates; the
{}placeholder in the template is replaced with the feature type.- Parameters:
Note
This method is experimental and is used by the Sequana RNA-seq pipeline to convert input GFF files to GTF format for RNA-seqc.
- class GenBank(filename, skip_types=['biological_region'])[source]#
This class reads a Genbank file
This is kept for back compatibility but most code in Sequana will rely on
sequana.gff3.GFF3instead. So please, transform your genbank to GFF3 format if possible.gg = GenBank() gg.get_types()
- property features#
Utilities for GTF files
- class GTFFixer(filename)[source]#
Some GTF have syntax issues. This class should fix some of them
With exons, we want to have unique IDs.
We append the transcrpt version and ID to the exon ID
Utilities to manipulate FastA files
- class FastaGFFCorrection(fasta_file, vcf_file)[source]#
Class to correct FastA reference and its GFF given a set of variants
- ::
f = FastaGFFCorrection("input.fa", "input.vcf") f.fix_and_save_fasta("new.fa") f.fix_and_save_gff("new.fa", "input.gff3", "new.gff3")
You must call
save_fasta()beforesave_gff()to compute the shifts of positions found in the VCF file.The GFF corrections are made on the start and stop positions of all features found in the GFF. In addition, gene and CDS start and stop positions are also corrected for the change of start/stop codon implied by the variants.
- fix_and_save_fasta(outfile)[source]#
We consider that the VCF file contains variants that have already been filtered.
Todo
handle multiple-variants at a given position
- fix_and_save_gff(gff_infile, fasta_corrected, gff_outfile, maxshift=8000)[source]#
Corrects the start/stop codons of input GFF given corrected fasta
You must call
fix_and_save_fasta()first.Todo
multiple contig is not yet implemented
- class Aragorn[source]#
# Example usage:
file_path = 'aragorn_output.txt' # Replace with the path to your Aragorn output file parsed_results = parse_aragorn_output(file_path) for result in parsed_results: print(result)
- class RNAmmer(filename)[source]#
input is the GFF file from rnammer that is in gff2 format.
##gff-version2 ##source-version RNAmmer-1.2 ##date 2024-09-09 ##Type DNA # seqname source feature start end score +/- frame attribute # --------------------------------------------------------------------------------------------------------- 11 RNAmmer-1.2 rRNA 394875 394989 59.3 + . 8s_rRNA 21 RNAmmer-1.2 rRNA 605807 605921 60.0 + . 8s_rRNA 9 RNAmmer-1.2 rRNA 417374 417488 63.6 + . 8s_rRNA 21 RNAmmer-1.2 rRNA 319355 319469 61.1 - . 8s_rRNA
VCF#
Python script to filter a VCF file
- compute_fisher_strand_filter(record)[source]#
Fisher strand filter (FS).
Sites where the numbers of reference/non-reference reads are highly correlated with the strands of the reads. Counting the number of reference reads on the forward strand and on the reverse strand, and the number of alternate reads on the forward and reverse strand should be equivalent. With these four numbers, we construct a 2 x 2 contingency table and used the P-value from a Fisher’s exact test to evaluate the correlation.
from sequana import freebayes_vcf_filter v = freebayes_vcf_filter.VCF_freebayes("data/JB409847.vcf") r = next(v) compute_fisher_strand_filter(r)
If the pvalue is less than 0.05 we should reject the variant since the alternate and reference do not behave in the same way. Typically found if the alternate has a poor strand balance. Used with care if frequency of alternate or reference is low or deth of coverage is low.
Note
fisher terst with two-sided hypothesis
- compute_frequency(record)[source]#
- Compute frequency of alternate allele.
alt_freq = Count Alternate Allele / Depth
- Parameters:
record -- variant record
- compute_strand_balance(record)[source]#
Compute strand balance of alternate allele include in [0,0.5]. strand_bal = Alt Forward / (Alt Forward + Alt Reverse)
- Parameters:
record -- variant record
FYI: in freebayes, the allele balance (reported under AB), strand bias counts (SRF, SRR, SAF, SAR) and bias estimate (SAP) can be used as well for filtering. Here, we use the strand balance computed as SAF / (SAF + SAR)
- strand_ratio(number1, number2)[source]#
Compute ratio between two number. Return result between [0:0.5].
- class FilteredVariantFile(variants, fb_vcf)[source]#
Variants filtered with VCF_freebayes.
Class kept for back compatiblity in wrappers and variant_calling pipeline
constructor
- Parameters:
variants (list) -- list of variants record.
fb_vcf (VCF_freebayes) -- class parent.
- property df#
Get the data frame.
- to_csv(output_filename, info_field=False)[source]#
Write DataFrame in CSV format.
- Params str output_filename:
output CSV filename.
- to_vcf(output_filename)[source]#
Write VCF file in VCF format.
- Params str output_filename:
output VCF filename.
- property variants#
Get the variant list.
- property vcf#
Get the VCF_freebayes object.
- class VariantFile(filename, progress=False, keep_polymorphic=True)[source]#
Reads a BCF or VCF or GZIPPED VCF
v = VariantFile()
You can access to a list of all variants (unchanged from pysam) using:
vlist = v.variants
you may transform a variant into a dictionary that contains a subset of most important information including SNPeff annotation (if available) using:
v._variant_to_dict(vlist[0])
A dataframe of all variants processed with this _variant_to_dict function is provided as an attribute:
v.df
Samples and contigs/chromosomes are available also as attributes:
v.samples v.contigs v.chromosomes # a synomym of v.contigs
There is a set of parameter to filter variants. To apply it, use the filter_vcf function.
fvcf = v.filter_vcf()
- property chromosomes#
- property contigs#
- property df#
- filter_vcf(filter_dict=None)[source]#
Filter variants in the VCF file.
- Parameters:
filter_dict (dict) -- dictionary of filters. It updates the attribute
VCF_freebayes.filter_params
Return Filtered_freebayes object.
- property is_joint#
- manhattan_plot(chrom_name=None, bins=200, types=['ins', 'del', 'mnp', 'snp', 'complex', 'INS', 'DEL'])[source]#
- property samples#
- to_vcf(output_filename)[source]#
Write VCF file in VCF format.
- Params str output_filename:
output VCF filename.
- property variants#
Other formats#
- class PfamDomtblout(filepath)[source]#
parser for the output of hmmscan (domtblout format)
hmmscan --cpu 8 --domtblout pfam.domtblout Pfam-A.hmm temp.faa > pfam.hmmscan.txt
IEM class
- class BCLConvert(filename)[source]#
Reader and validator of Illumina v2 (BCL Convert) sample sheets.
BCL Convert is the replacement for the bcl2fastq software. The sample sheet format (a.k.a. v2) is close to the bcl2fastq one (a.k.a. v1) handled by
SampleSheetbut differs in a few places:The tabular data lives in a
[BCLConvert_Data]section (required) instead of[Data].Settings live in a
[BCLConvert_Settings]section (required).[Settings]may still appear but is optional.[Reads]uses cycle counts (Read1Cycles,Index1Cycles, ...) rather than one integer per line.The header carries
FileFormatVersion,2.Sample_IDis compulsory (error if absent) and at least one sample is required.Sample_Nameis ignored by BCL Convert (warning if present).Adapter trimming/masking options moved into the settings section, e.g.
AdapterbecameAdapterRead1and command-line options such as--minimum-trimmed-read-lengthbecameMinimumTrimmedReadLength.Barcode mismatches are set in the settings with
BarcodeMismatchesIndex1andBarcodeMismatchesIndex2.
Because sections are parsed generically by
SampleSheet._scan_sections(), most of the [Data] checks (mandatory columns, unique Sample_ID, unique/valid indices, homogeneous index lengths, ...) are reused as-is by pointingdata_sectionandsettings_sectionto the v2 sections.- References:
- data_section = 'Bclconvert_Data'#
the tabular section is [BCLConvert_Data]; title-cased by the parser
- quick_fix(output_filename)[source]#
Fix a v2 sample sheet by removing trailing semicolons only.
Unlike
SampleSheet.quick_fix(), internal semicolons are preserved because they are legitimate v2 separators (e.g.OverrideCyclesusesR1:Y151;I1:I10;I2:I10;R2:Y151). Only trailing;(typical Excel artefact) are stripped.
- settings_section = 'Bclconvert_Settings'#
the settings section is [BCLConvert_Settings]; title-cased by the parser
- class SampleSheet(filename)[source]#
Reader and validator of Illumina samplesheets
The Illumina samplesheet reader and validator verifies the correctness of the sections in the samplesheet, which are not case-sensitive and are enclosed within square brackets.
Following the closing bracket, no additional characters are permitted, except for commas and the end-of-line marker. For instance [Data]a prevents the [Data] section from being correctly processed.
The sections then consist of key-value pairs represented as records, with each line consisting of precisely two fields.
An optional [Settings] section can contain key-value pairs, and the [Reads] section specifies the number of cycles per read, which is exclusively required for MiSeq.
The [Data] section, which is a table similar to CSV format, is optional. However, without [Data] section all reads are sent to a single 'undetermined' output file. Sample_ID is highly recommended.
Example of typical Data section to be used with bcl2fastq:
[Header] [Data] Sample_ID,Sample_Name,I7_Index_ID,index,I5_INdex_ID,index2 A10001,Sample_A,D701,AATACTCG,D501,TATAGCCT A10002,Sample_B,D702,TCCGGAGA,D501,TATAGCCT A10003,Sample_C,D703,CGCTCATT,D501,TATAGCCT A10004,Sample_D,D704,GAGATTCC,D501,TATAGCCT
Important: altough we have upper case names as specified in the Illumina specs, the bcl2fastq does not care about the upper case. This is not intuitive since IEM produces keys with upper and lower case names similarly to the specs.
Sequana Standalone
The standalone application sequana contains a subcommand based on this class:
sequana samplesheet
that can be used to check the correctness of a samplesheet:
sequana samplesheet --check SampleSheet.csv
- References:
illumina specifications 970-2017-004.pdf
- data_section = 'Data'#
name of the section holding the tabular sample data (title-cased). Overridden in
BCLConvertwhere the section is[BCLConvert_Data].
- property df#
Returns the [Data] section
- expected_data_headers = {'PE': [], 'SE': []}#
- expected_headers_fields = ['IEMFileVersion', 'Investigator Name', 'Instrument Type', 'Experiment Name', 'Date', 'Workflow', 'Application', 'Assay', 'Description', 'Chemistry', 'Index Adapters']#
- property header#
- property index_adapters#
returns index adapters
- property instrument#
returns instrument name
- quick_fix(output_filename)[source]#
Fix sample sheet
Tyical error is when users save the samplesheet as CSV file in excel. This may add trailing ; characters at the end of section, which raises error in bcl2fastq.
- property samples#
returns the sample identifiers as a list
- property settings#
- settings_section = 'Settings'#
name of the settings section (title-cased). Overridden in
BCLConvertwhere the required section is[BCLConvert_Settings].
- to_fasta(adapter_name='')[source]#
Extract adapters from [Adapter] section and print them as a fasta file
- validate()[source]#
This method checks whether the sample sheet is correctly formatted
- Checks for:
presence of ; at the end of lines indicated an edition with excel that wrongly transformed the data into a pure CSV file
inconsistent numbers of columns in the [DATA] section, which must be CSV-like section
Extra lines at the end are ignored
special characters are forbidden except - and _
checks for Sample_ID column uniqueness
checks for index uniqueness (if single index)
checks for combo of dual indices uniqueness
checks that sample names are unique
and raise a SystemExit error on the first found error.
- property version#
return the version of the IEM file
- SampleSheetFactory(filename)[source]#
Return a
BCLConvertorSampleSheetinstance based on the detected sample sheet version (seeget_sample_sheet_version()).
- get_sample_sheet_version(filename)[source]#
Return
"v2"for BCL Convert sample sheets,"v1"otherwise.Detection relies on the v2 markers: presence of a
[BCLConvert_Data]or[BCLConvert_Settings]section, orFileFormatVersion,2in the header.
- class Cigar(cigarstring: str)[source]#
A class to handle CIGAR strings from BAM files.
>>> from sequana.cigar import Cigar >>> c = Cigar("2S30M1I") >>> len(c) 33 >>> c = Cigar("1S1S1S1S") >>> c.compress() >>> c.cigarstring '4S'
Possible CIGAR types are:
"M" : alignment match
"I" : insertion to the reference
"D" : deletion from the reference
"N" : skipped region from the reference
"S" : soft clipping (clipped sequence present in seq)
"H" : hard clipping (sequence NOT present)
"P" : padding (silent deletion from padded reference)
"=" : sequence match
"X" : sequence mismatched
"B" : back (rare) (could be also NM ?)
!!! BWA MEM get_cigar_stats returns list with 11 items Last item is !!! what is the difference between M and = ??? Last item is I + S + X !!! dans BWA, mismatch (X) not provided... should be deduced from last item - I - S
Note
the length of the query sequence based on the CIGAR is calculated by adding the M, I, S, =, or X and other operations are ignored. source: https://stackoverflow.com/questions/39710796/infer-the-length-of-a-sequence-using-the-cigar/39812985#39812985
- Reference:
Constructor
- Parameters:
cigarstring (str) -- the CIGAR string.
Note
the input CIGAR string validity is not checked. If an unknown type is found, it is ignored generally. For instance, the length of 1S100Y is 1 since Y is not correct.
- as_dict()[source]#
Return cigar types and their count
- Returns:
dictionary
Note that repeated types are added:
>>> c = Cigar('1S2M1S') >>> c.as_dict() {"S":2,"M":2}
- as_tuple()[source]#
Decompose the cigar string into tuples keeping track of repeated types
- Returns:
tuple
>>> from sequana import Cigar >>> c = Cigar("1S2M1S") >>> c.as_tuple() (('S', 1), ('M', 2), ('S', 1))
- property cigarstring#
- pattern = re.compile('(\\d+)([A-Za-z])?')#
- stats()[source]#
Returns number of occurence for each type found in
types>>> c = Cigar("1S2M1S") >>> c.stats() [2, 0, 0, 0, 2, 0, 0, 0, 0, 0]
- types = 'MIDNSHP=XB'#
Sniffer