8. Sequanix Tutorial#
8.1. Quick Installation#
Sequanix is shipped with Sequana. If you use conda, just type:
conda install sequana
Please see Installation for details. The standalone name is sequanix.
8.2. Introduction#
Sequanix is a graphical user interface (GUI) that can be used to run Snakemake workflows. The standalone name is sequanix (small caps) and is part of Sequana library.
The primary goal was to provide a GUI to easily run Sequana pipelines (designed as Snakemake workflows).
However, we extended the interface so that it can handle other Snakemake workflows, referred to as Generic pipelines in the GUI.
A tutorial in form of a video is available in another repository: video/tutorial
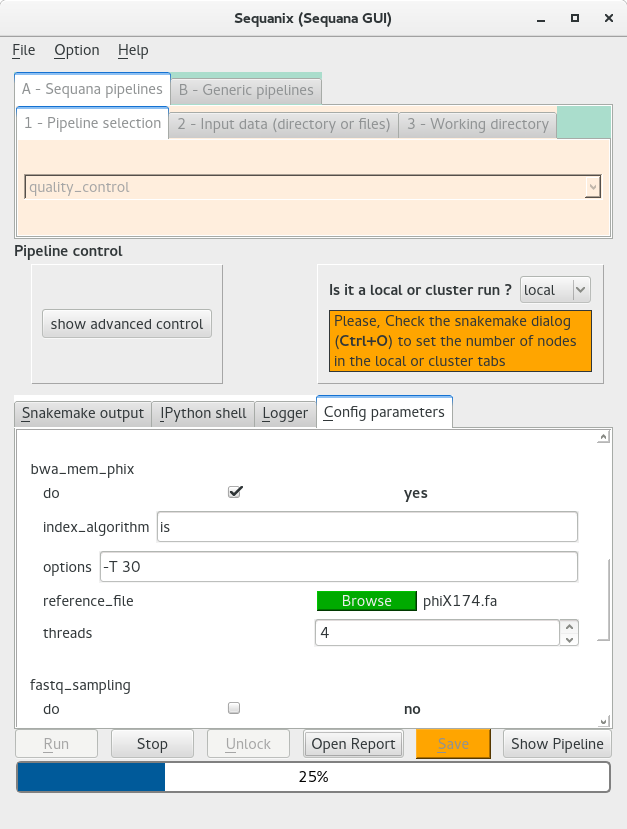
Snapshot of the Sequanix graphical user interface (GUI)#
In this section, we first show how to run one of our Sequana pipeline (quality control pipeline). Second, we show how to run Generic pipelines that are not part of Sequana. For these two examples, the computation is done locally. However, one strength of Snakemake pipelines is that they can be executed on various cluster without changing the pipeline itself. This is also possible via Sequanix as explained in the Running analysis on a cluster section (SLURM and SGE frameworks).
Snakemake pipelines are made of 2 parts: a pipeline and an optional configuration file; The pipeline may be called Snakefile. It contains the code of the pipeline itself. Keep in mind that in the Snakefile, developer may link the pipeline to an external configuration file: the config file, which is encoded in YAML or JSON format.
8.3. Sequana pipeline: the quality control example#
8.3.1. Prerequisites: get some data#
The following example will show how to run a quality control pipeline on a pair of FastQ files. The data comes from a sequencing platform (using HiSeq technology) of a Measles virus. For testing purposes, you can download R1 and R2)
files that contain only 1500 reads. Copy the two files in a local directory (let us call
it testing) and start sequanix.
cd testing
sequanix
8.3.2. Select the quality control pipeline#
First you need to select the pipeline of interest (here the quality_control). In the following figure, you need to
select the sequana pipelines tab (arrow 1),
select the pipeline section tab (arrow 2)
select the pipeline in the dropdown box (arrow 3)
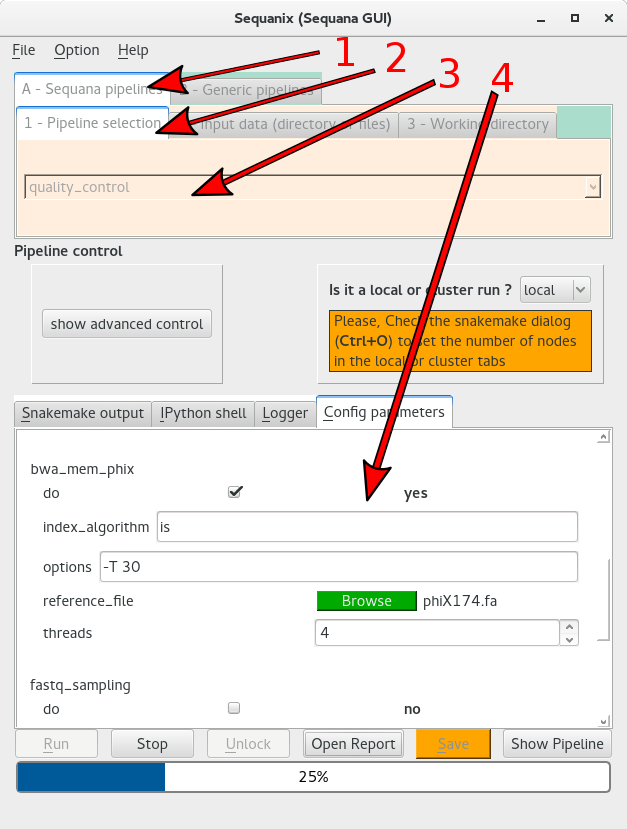
Once done, the configuration file of the pipeline will be loaded in the Config parameters tab (arrow 4).
If a file named config_cluster.josn is defined by the developer in the pipeline module, then it is loaded automatically in the snakemake dialog and used if the pipeline is executed on a cluster.
8.3.3. Select the input data (directory or files)#
Once the pipeline is selected, you need to give information about the location of the input data. Generally, the pipelines take fastq.gz files as input.
In general we have many samples, so you need to select the Input directory tab. If you have only one or two files, you may use the other tab (Input sample(s)).
Here, we consider the first case only (directory). First click on the red Browse button (figure below) to select the directory where is stored the data.
By default, we assume that there is a special tag in the filenames (_R1_ or _R2_) but one can change it to another pattern. Note also that we expect by default the input files to end up in fastq.gz
So by default if you select a directory, all files ending in fastq.gz will be selected.
Note
if FastQ files are stored in various directories, use the Optional pattern. For instance if samples are stored in sub directories, use */*fastq.gz
Note
The directory browser can create directories
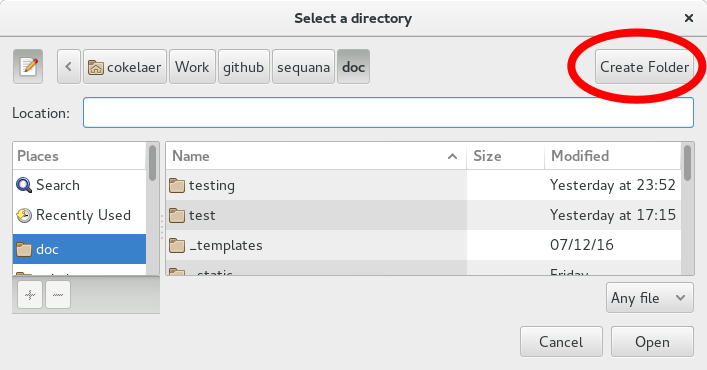
In the browser, you can either select an existing directory, or create a new one by clickin on the Create Folder (red ellipse on top left corner). Note that depending on your system, the layout may be different#
8.3.4. Select the working directory#
As above, when clicking on the browser button, you may select an existing one or create a new one.
8.3.5. Fine tune the config parameters#
Warning
Sequana pipelines may be complex with several dependencies on external tools. We would recommend users to look at the online documentation for help (e.g., Tutorial, Pipelines).
One major interest of Sequanix is that the Snakemake configuration file is loaded and can then be changed dynamically. In other word, you do not need to use an esoteric text editor, which may be the only option on a cluster.
Moreover, the loaded configuration file has other advantages:
file can be selecting thanks to a file browser. If no file is selected, the button is red (green otherwise)
Some buttons have dedicated widgets (e.g. in the figure above, the number of threads has its own dropbox limited typing errors)
Boolean fields have their own checked buttons
etc
Note
For developers: please see the Documenting the configuration file section to see how to write your configuration to have the widgets loaded automatically.
8.3.6. Save, check and run the project#
Once the parameters have been set, it is time to save the project. You can either click the yellow box Save in the bottom bar or the Ctrl+S shortcut.
The configuration and pipelines files are then save in the working directory defined above. If the files already exists, a dialog box ask you to confirm that you want to overwrite the existing files.
You can then check the pipeline by clicking the Show Pipeline button or use Ctrl+D shortcut. For simple pipeline, this may not be very useful but for complex dynamic pipelines where parts may be switched off, this may be convenient.
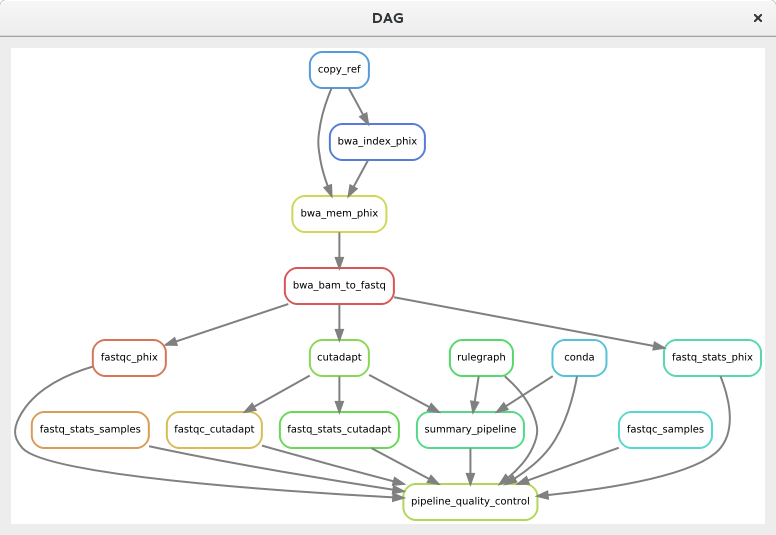
A dialog showing the DAG (directed acyclic graph) with dependencies in the analysis pipeline#
Finally, once saved, the Run button should be clickable. Click on it or use Ctrl+R shortcut. The output of Snakemake will be shown and the progress bar will move showing the stage of the analysis.
Warning
with long analysis, the progress bar may be stalled for a while. It may even stay at 0% for a long time. Just be patient.
8.3.7. Stopping a running analysis:#
If you realise that you made a mistake in the configuration or simply want to stop the current analysis, click the Stop button
8.3.8. Start Sequanix with pre-defined values#
If you use Sequanix regularly, it may be convenient to start the standalone with with pre-filled values. For instance, to pre-fill the input directory, the working directory and the pipeline itself start Sequanix as follows:
sequanix -i . -p quality_control -w analysis
For help, please type:
sequanix --help
8.4. Generic pipeline: a minimalist example with no configuration file#
In this section we will use a very simple Snakefile that reads FastQ files (gzipped) and counts the number of reads (not lines). The results are then summarised into a file named summary.txt. For those who are curious, here is the Snakefile.
1import glob
2import os
3
4# Input parameter to be changed
5directory = "."
6
7directory = os.path.abspath(directory) + "/"
8filenames = glob.glob(directory + "*.fastq.gz")
9samples = [os.path.split(this)[1].replace(".fastq.gz", "")
10 for this in filenames]
11
12rule all:
13 input: "summary.txt"
14
15rule summary:
16 input: expand("count_{sample}.txt", sample=samples)
17 output: "summary.txt"
18 shell: "cat count_*.txt > summary.txt"
19
20rule count:
21 input: directory + "{sample}.fastq.gz"
22 output: temp("count_{sample}.txt")
23 shell: "gunzip -c {input} | wc - | awk '{{print $1/4}}' > {output}"
Note
In this example, the directory where to find the data is hardcoded so you must change it (see highlighted line in the code below).
This example does not depend on any external configuration file. We will see later on to combine this Snakefile with a configuration file where the directory can be set.
8.4.1. Prerequisites: get some FastQ files#
To run the pipeline, we first need to get some FastQ files (zipped). We need to store them in a data directory. Create one and move into the directory as follows:
mkdir data
cd data
If you do not have FastQ files, get the following ones:
You will also need the Snakefile (pipeline) itself:
Warning
if the data and pipeline are in a different directories, you need to change the highlighted line (line 5) to set the directory name specifically.
Once ready, start Sequanix in a shell:
sequanix
8.4.2. The analysis#
Similarly to the Sequana pipeline case, you need to select the pipeline as follows:
Select the Generic pipelines tab (arrow 1)
Select the pipeline section tab (arrow 2)
Click on the browse button to select the pipeline file (minimalist.rules)
There is no configuration file so we can now save and run the project:
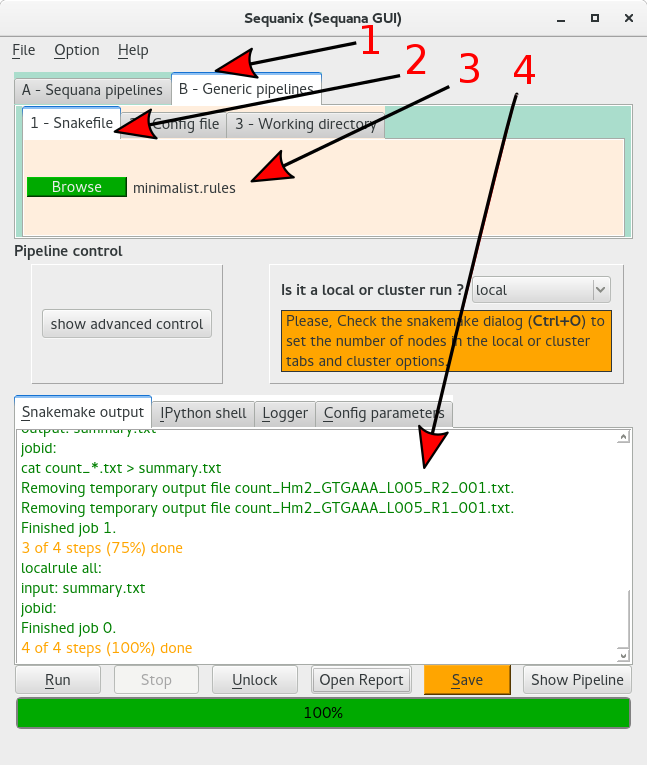
8.4.3. Save, check and run the project#
Same as for a Sequana pipeline
8.4.4. Start Sequanix with pre-defined values#
Alternative way to start sequanix with pre-filled values for the working directory and the pipeline file:
sequanix -w analysis -s minimalist.rules
8.5. Generic pipeline: a minimalist example with a configuration file#
In this section, we use a pipeline that is almost identical to the previous one.
1import glob
2import os
3
4configfile: "minimalist.yaml"
5directory = config['data_directory']
6
7directory = os.path.abspath(directory) + "/"
8filenames = glob.glob(directory + "*.fastq.gz")
9samples = [os.path.split(this)[1].replace(".fastq.gz", "")
10 for this in filenames]
11
12rule all:
13 input: "summary.txt"
14
15rule summary:
16 input: expand("count_{sample}.txt", sample=samples)
17 output: "summary.txt"
18 shell: "cat count_*.txt > summary.txt"
19
20rule count:
21 input: directory + "{sample}.fastq.gz"
22 output: temp("count_{sample}.txt")
23 shell: "gunzip -c {input} | wc - | awk '{{print $1/4}}' > {output}"
The only difference is on line 4 and 5: the previously hard-coded variable directory is now extracted from an external configuration file called minimalist.yaml.
Here are the links to get the Snakefile and the configuration file.
Similarly to the previous example you would need some FastQ files (see Prerequisites: get some FastQ files). Once done, start sequanix. Here you would need to load the pipeline and set the working directory but also to load the config file. When you load the config file, you should see something equivalent to the following figure: the configuration file is shown in the Config parameters section:
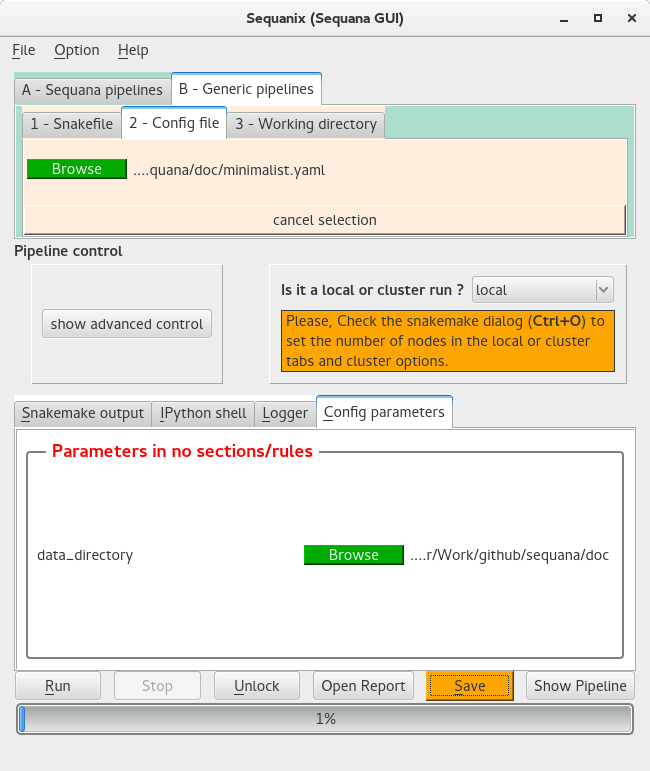
You can see here that the configuration file (a single parameter data_directory) is interpreted and a widget is available to select the directory where to find the data (for developers, please see Developer guide section).
The rest of the analysis works as above.
8.6. Dialogs and running analysis locally or on a cluster#
So far we have used Sequanix with the default parameters.
8.6.1. The Sequanix browser and the preferences dialog#
8.6.1.1. sequana_fox, the sequanix home-made browser#
Once an analysis is finished, Sequana pipeline generally creates an HTML report. This is the reason why we added an Open Report button in the bottom. This opens a file browser where users can select an HTML file. The browser used by default is a home-made browser so that it can run on a cluster where no standard browser are installed.
The home-made browser, which can be used as a standalone (sequana_fox) is simple but should be enough for most HTML pages. There is a forward/backward capability, support for Javascript, ability to change the URL but that is pretty much all. This is mainly used to check that HTML files have been created.
8.6.1.2. Preferences dialog#
In order to open the preferences dialog, type Ctrl+P or go to the Option menu at the top and select Preferences. The Preferences dialog looks like the following figure:
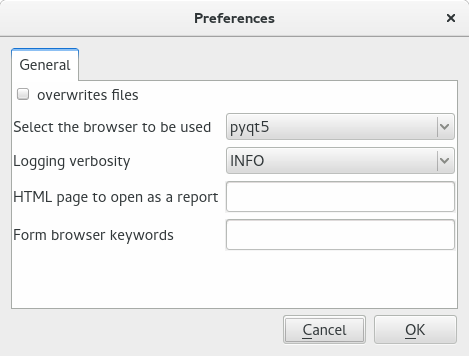
Preferences dialog. This dialog is accessible via the menu or the short Ctrl+P. It contains general options to tune Sequanix's behaviour#
Brief description of the options:
- overwrite files:
if checked, when saving a project, the existing configuration and pipelines are overwritten
- select the browser to be used:
By default the home-made browser (pyqt5) is used but one can select firefox, safari, chrome instead.
- logging verbosity:
there are 5 level of verbosity. By default, we use INFO. It may be useful to set the option to DEBUG if there are errors and you wish to provide a complete bug report to Sequana developers.
- HTML page to open as a report:
If you set a filename here, then when pressing Open report, instead of opening a file browser, sequanix open the file provided.
- Form browser keywords:
In the config parameters, if you wish to associate a parameter name with a browser widget, add the names here (separated by comma)
8.6.2. From a local to cluster analysis#
One strength of Snakemake (and Sequanix) is that pipelines can be run locally but also on clusters using various scheduler frameworks without changing the pipeline code.
In Sequanix, we can switch between a local run or a cluster run by switching a button in the main window as shown in the figure below:
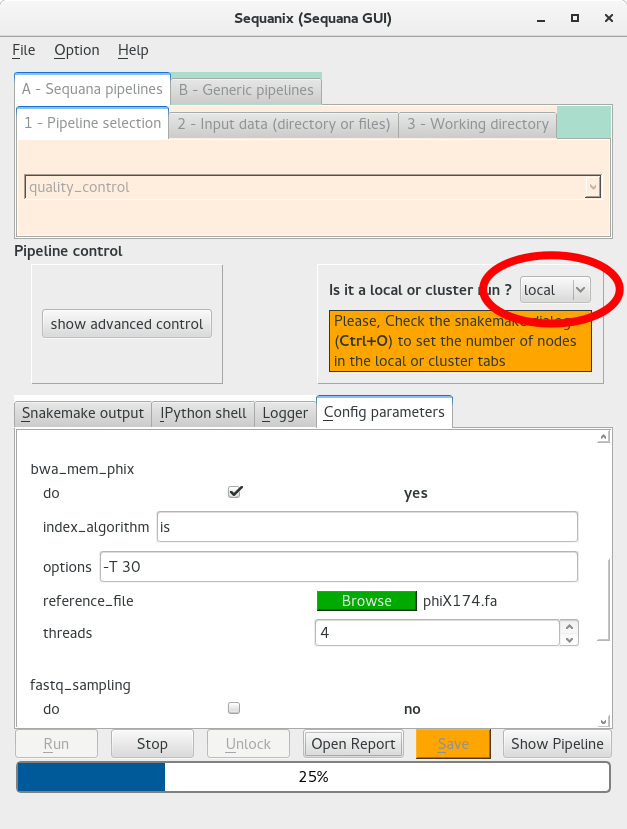
If you are on a cluster, you should switch the local mode to cluster AND you have to provide the cluster commands in the Snakemake dialog (see above).#
The Snakemake dialog contains 3 sub tab: the local, cluster and general tabs.
8.6.2.1. Running analysis locally#
If you run the analaysis locally, you do not need to change much. The only option to tune is the number of cores to be used locally. This happens in the Local tab. By default the cores parameter is set to the number of cores found on the computer. You may reduce this number if you wish.
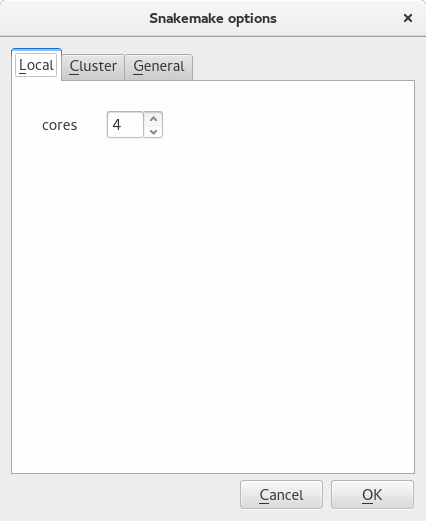
The local tab contains only one option to set the number of local cores to be used. By default it is the number of available cores on the machine used.#
8.6.2.2. Running analysis on a cluster#
If you run the analysis on a cluster, this is a bit more complicated.
First, similarly to the local run, you may provide the number of cores to be used. This happens in the Cluster tab. Here, you can set the parameters jobs to the required number of CPUS. If you know that at a given time, you may have N jobs running, set this parameter to N. For instance, if you have 48 samples, and you perform 48 independent analysis, set jobs to 48.
Second, you must set the cluster commands. We will not provide an exhaustive documentation on this aspect, which is technical and pipeline and cluster dependent.
We provide two examples. First, let us assume the case where :
you are on a cluster with a SLURM framework
your jobs require less than 4 Gb of memory
Then, you must add this line in the cluster field
sbatch --mem=4000
Second, let us assume the case where:
you are on a cluster with a SGE framework
your jobs require 4 threads each
Then, you must add this line in the cluster field
qsub --pe threaded 4
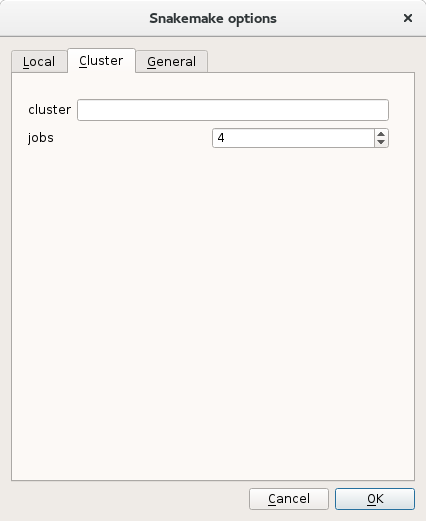
The tab cluster contains parameters related to the execution of the Snakemake pipeline can be set (e.g. specific job scheduler information or number of CPUs to be used).#
8.6.2.3. Other Snakemake options#
Snakemake itself has lots of options. In the snakemake dialog, in the General tab, one can set them.
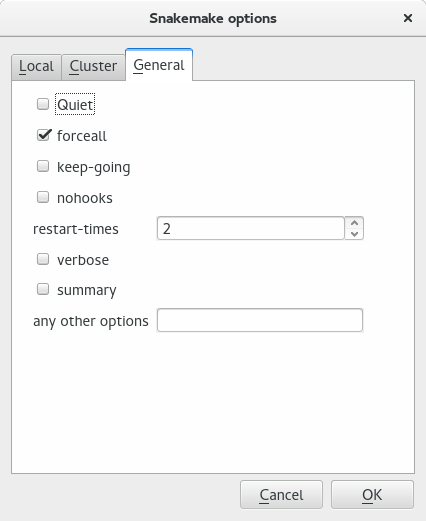
In the General tab, check boxes related to Snakemake are available. Any other options can be set in the editable line at the bottom.#
Here is a brief description:
- quiet:
Do not output any progress or rule information
- forceall:
Force the execution of the selected (or the first) rule and all rules it is dependent on regardless of already created output.
- keep-going:
Go on with independent jobs if a job fails.
- nohooks:
Do not invoke onstart, onsuccess or onerror hooks after execution.
- restart-times:
Number of times to restart failing jobs (defaults to 0).
- verbose:
Print debugging output
- summary:
Print a summary of all files created by the workflow. The has the following columns: filename, modification time, rule version, status, plan. Thereby rule version contains the version the file was created with (see the version keyword of rules), and status denotes whether the file is missing, its input files are newer or if version or implementation of the rule changed since file creation. Finally the last column denotes whether the file will be updated or created during the next workflow execution.
- any other options:
8.7. FAQS#
8.7.1. How to run Sequanix on a SLURM cluster.#
You have to connect with ssh and the -X option:
ssh -X your.cluster.address
Once connected, on a slurm system type:
srun --x11 sequanix
8.7.2. What to do if a RUN fails#
An analysis may fail for various reasons. The errors have several origins. Most commons ones are:
configuration file not filled properly (e.g. missing input file)
Input data not found (e.g., bad pattern)
bug in the pipeline (Fill an issue on http://github.com/sequana/sequana/issues)
bug in Sequanix (fill an issue)
cluster issue: a job is killed because not enough memory was allocated (adapt the cluster option by increasing relevant resources such as memory requirements)
By experience, the first 2 type of errors are the most common.